Leggo frequentemente nelle schede tecniche di alcune specialità di una proteina di trasporto, chiamata glicoproteina-P, coinvolta nella cinetica e citata soprattutto in relazione alle interazioni tra i farmaci. A cosa serve? Perché è importante?
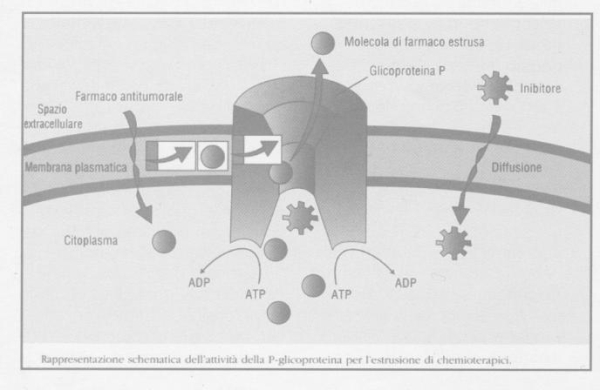
La glicoproteina-P, anche nota come ABCB1, è il componente meglio conosciuto di una famiglia di proteine di trasporto, chiamate ABC, che impiegano l'energia derivante dall'idrolisi dell'ATP per trasportare molecole attraverso la membrana cellulare. La glicoproteina-P trasporta le molecole all'esterno della cellula ed è pertanto classificata come una pompa di efflusso. L'interesse nei confronti di questa proteina è iniziato quando si è intuito che la sua iperespressione nelle cellule tumorali causava una forma di resistenza multipla ad alcuni tipi di chemioterapici (es. paclitaxel). Oggi è accertato che questa proteina sia espressa anche in molti tessuti normali e che l'espressione costitutiva della glicoproteina-P sia, al pari degli enzimi metabolizzanti, un importante meccanismo protettivo contro la potenziale tossicità di sostanze esogene, farmaci inclusi. In base alla sua localizzazione anatomica, si possono identificare 3 funzioni:
1. limitazione dell'assorbimento del farmaco: espulsione del farmaco, appena assorbito dagli enterociti (cellule della parete intestinale), nel lume intestinale ed eliminazione con le feci.
2. eliminazione attiva del farmaco: a) trasferimento del farmaco dalle cellule del tubulo prossimale al lume tubulare ed eliminazione del farmaco con le urine; b) trasferimento del farmaco dagli epatociti alla bile.
3. limitazione della distribuzione del farmaco ai tessuti: ad esempio trasferimento del farmaco al di fuori della barriera emato-encefalica (riducendo così gli effetti indesiderati di tipo neurologico di alcuni farmaci, ma anche limitando l'efficacia di altri).
Il ruolo della glicoproteina-P nell'interazione tra farmaci
Analogamente a quanto accade con gli enzimi epatici del citocromo P450, anche la glicoproteina-P può essere indotta o inibita da alcuni farmaci portando ad una riduzione o ad un aumento della sua capacità di efflusso. L'influenza dei farmaci sulla glicoproteina-P, così come per il citocromo P450, è alla base di interazioni di tipo farmacocinetico tra farmaci. La rilevanza di questa interazione dipende dalla localizzazione e dalla funzione della glicoproteina-P alterata.
Così ad esempio:la mancanza di effetti indesiderati della loperamide a livello del sistema nervoso centrale sembra dovuta alla sua espulsione da parte della glicoproteina-P; in caso però di contemporanea assunzione di chinidina, che inibisce questo efflusso, alcuni pazienti possono sviluppare effetti avversi di tipo neurologico come risultato di un'aumentata esposizione alla loperamide;
la clearance renale e biliare della digossina è mediata dalla glicoproteina-P di cui il verapamil è un inibitore. La contemporanea assunzione di verapamil riduce l'escrezione della digossina aumentandone la biodisponibilità e potrebbe rendersi necessario ridurre il dosaggio di digossina per prevenire effetti tossici.
Non sempre l'interazione ha rilevanza clinica. Ad esempio la rifampicina induce la glicoproteina-P intestinale, portando ad una riduzione dell'assorbimento di digossina e ad una modesta diminuzione dei livelli ematici che generalmente non si traduce in effetti clinici rilevanti. La glicoproteina-P è capace di trasportare una notevole varietà di composti strutturalmente diversi tra loro, molti dei quali sono anche i substrati del'CYP3A4, un isoenzima della famiglia del citocromo P450 che metabolizza i farmaci. Alcune evidenze suggeriscono che la glicoproteina-P e il CYP3A4 operino in maniera coordinata sull'esito complessivo delle interazioni farmacologiche, dal momento che molti farmaci substrati della glicoproteina-P lo sono anche del CYP3A4. Così, se un farmaco che inibisce sia la glicoproteina-P che il CYP3A4 intestinale viene assunto contemporaneamente ad un farmaco substrato di entrambi i sistemi, la biodisponibilità di quest'ultimo sarà maggiore sia per la riduzione della quota espulsa a livello intestinale dalla glicoproteina-P che per la riduzione del metabolismo ad opera del CYP3A4.
L'effetto "netto" delle interazioni che coinvolgono glicoproteina-P e il CYP3A4 dipenderà quindi dall'affinità relativa del farmaco per l'uno o l'altro sistema e dall'entità dell'inibizione o dell'induzione provocata. Questa sinergia potrebbe essere alla base di molte interazioni farmacocinetiche che non sono ancora ben chiare. Ad esempio sia il verapamil che il diltiazem sono considerati moderati inibitori del CYP3A4, ma il verapamil causa un aumento della biodisponibilità del sirolimus considerevolmente maggiore rispetto al diltiazem. Il verapamil è un noto inibitore anche della glicoproteina-P, mentre il diltiazem non sembra influenzarne l'attività, quindi il maggiore effetto del verapamil sui livelli di sirolimus si potrebbe spiegare per il duplice effetto inibitorio sia sulla glicoproteina-P che sul CYP3A4. Come si può immaginare, le implicazioni sulla cinetica dei farmaci sono complesse e molti aspetti sono tuttora oggetto di studio. Solo la ricerca potrà aiutarci a comprendere il ruolo complessivo di questa proteina nelle interazioni tra farmaci e soprattutto quale rilevanza clinica esse abbiano.
Bibliografia di riferimento
-P-glycoprotein: why is it significant? Learning & development. Pharmaceutical Journal (Vol 286) 21 May 2011. www.pjonline.com.
-Importanza funzionale della glicoproteina-P nella barriera sangue/tessuto e nelle interazioni tra farmaci. www.farmacovigilanza.org/corsi/050430-05.asp.
-Fromm MF. et al. Inhibition of P-glycoprotein-mediated drug transport: a unifying mechanism to explain the interaction between digoxin and quinidine. Circulation 1999; 99:552-57.
-Greiner B. et al. The role of intestinal P-glycoprotein in the interaction of digoxin and rifampin. J Clin Invest 1999; 104:147-53.
-Lin J. et al. H.Drug-drug interaction mediated by inhibition and induction of P-glycoprotein. Adv Drug Deliv Rev 2003;55:53-81.
-Sadeque AJ. et al. Increased drug delivery to the brain by P-glycoprotein inhibition. Clin Pharmacol Ther 2000; 68:231-37.


{kind=link}