

Nota redazionale Dedicare un aggiornamento di IsF ad un tema oggettivamente molto specialistico come quello annunciato nel titolo con la sua dizione ufficiale sembrerebbe rompere una tradizione che cerca di mantenere un’attenzione privilegiata per interessi più generali dei lettori. Non è difficile tuttavia ricordarsi, una volta che il titolo viene ricondotto al nome comune - cellule staminali - con cui questa area di ricerca di base e clinica è passata dall’ambito strettamente scientifico agli immaginari individuali e collettivi, per rendersi conto che il tema è assolutamente ed esemplarmente pertinente: per i suoi contenuti, e per quello che significa a livello di strategie di informazione. Si tratta di fatto di un’area modello del rapporto che si instaura tra scienza e società quando all’impotenza della medicina nel rispondere a bisogni inevasi corrisponde l’intreccio (inevitabile?) di:
• tempi lunghi oggettivamente necessari per passare da un’ipotesi di risposta alla sua documentazione affidabile;
• la commistione ad alto rischio tra legittimità di attese ed emotività personali;
• la loro manipolazione da parte di comportamenti professionali scorretti;
• interessi economici “indipendenti” da qualsiasi criterio socialmente responsabile (etico è termine troppo “alto” in questo contesto);
• la incertezza-assenza dei poteri-saperi di controllo istituzionale.
Non è difficile riconoscere in questo scenario quanto è successo per il “Caso Stamina”, che evoca altri scenari, tipicamente (anche se non esclusivamente) italiani. L’aggiornamento è stato richiesto ai responsabili di un gruppoleader nel settore, che hanno scelto (e ne siamo felici) di offrirci una narrazione “complessiva” e realistica dei diversi scenari che si intrecciano per fare di questi ATMPs una risorsa terapeutica veramente affidabile. Può essere utile avere una conferma dell’interesse generale ed esemplare del tema in un numero recente del NEJM 1 . Il titolo della “Perspective” che ci interessa è talmente evocativo che non ha bisogno di commenti. Con la produzione-regolamentazione di ATMPs (su cui anche il nostro aggiornamento si concentra in chiusura) siamo, oggi, nel vecchio (mitico? caro? drammatico?) Wild West.
1. Taylor-Weiner H, Graff Zivin J: Medicine's Wild West - Unlicensed Stem-Cell Clinics in the United States N Engl J Med. 2015; 373:985-7
Cosa sono gli ATMPs e le loro differenze rispetto ai farmaci tradizionali Sempre più spesso in medicina viene proposto l’uso, con finalità terapeutiche, di cellule, geni, organi e tessuti, modificati e non. Tutto questo ampio spettro di proposte innovative viene classificato come “Advanced Therapeutic Medicinal Products” (ATMPs), ossia Prodotti Medicinali di Terapia Avanzata. Gli ATMPs differiscono molto dai farmaci tradizionali: in particolare il materiale di partenza è di origine biologica, generalmente umana (cellule o tessuti di donatori sani o di pazienti), e pertanto non è di facilissima reperibilità; a volte deriva dal paziente stesso (per trattamenti autologhi) o da un donatore compatibile in termine di istocompatibilità maggiore (HLA) (cioè il donatore delle cellule di partenza permette la produzione di un prodotto dedicato ad un singolo paziente o a pochi pazienti).
I Prodotti Medicinali di Terapia Avanzata (Advanced Medicinal Products, ATMPs) sono medicine per uso umano basate su geni o cellule. Sono farmaci innovativi per il trattamento di malattie o danni tessutali e possono essere classificati come:
1. Farmaci di Terapia Genica, quando contengono geni;
2. Farmaci di Terapia Cellulare Somatica, quando sono composti di cellule o tessuti manipolati in vitro al fine di cambiarne le proprietà biologiche;
3. Farmaci di Ingegnerizzazione dei Tessuti, quando contengono cellule o tessuti che possono rigenerare o riparare dei tessuti umani;
4. ATMPs combinati, che contengono uno o più dispositivi medici come parte integrante del farmaco, per esempio cellule poste su supporti inerti.
Inoltre qualsiasi materiale biologico umano mostra una grande variabilità da donatore a donatore, intrinseca alla diversità genetica, di età, sesso ecc. dei donatori di queste cellule. Anche la quantità del materiale di partenza è limitata e pertanto permette la produzione di lotti di prodotto relativamente piccoli dedicati a pochi pazienti, spesso orfani di alternative terapeutiche o con malattie rare. I farmaci tradizionali sono invece, nella maggior parte dei casi, il risultato di sintesi chimiche, o prodotti ricombinanti che possono essere generati da cloni di cellule molto omogenee tra di loro; possono essere prodotti su larga scala, per esempio utilizzando dei fermentatori. Forse le uniche eccezioni sono alcuni prodotti (molecole) purificati da organi animali o umani (ad esempio dal plasma di pool di donatori).
Gli ATMPs pertanto, in ragione della loro peculiarità, come descritto in precedenza, sono stati quasi sempre sviluppati (come fase preclinica fino agli studi clinici di Fase I/II) in ambiti accademici o ospedalieri e di ricerca da parte di medici/biologi/ricercatori fortemente coinvolti nella prospettiva di “tradurre” in clinica i risultati delle scienze più sperimentali (ad esempio in immunologia, ematologia ed oncologia).
Per trapianto di cellule staminali ematopoietiche si intende la somministrazione ad un paziente (in genere ammalato di leucemia, linfoma o mieloma), che è stato totalmente deprivato della sua ematopoiesi (perché tumorale) a seguito di chemio- e radioterapia, di cellule staminali derivate dal sangue o dal midollo di un donatore sano geneticamente compatibile (trapianto allogenico). Ad alcune ore/giorni dalla somministrazione in vena, le cellule staminali si localizzano nelle “nicchie” ematomidollari e, col passar del tempo, ripopolano tutto il midollo e poi il sangue circolante con tutti gli elementi del sistema ematopoietico (globuli rossi, globuli bianchi, piastrine).
Inoltre, in questi stessi ambiti accademici ed ospedalieri, la recente storia di “progresso” aveva portato a sviluppare strategie di laboratorio gestibili nell’accademia, vedi come esempio ideale tutta la storia del trapianto di cellule staminali ematopoietiche ed i successivi costanti miglioramenti per trattare al meglio il trapianto e le sue complicanze, per cui le cellule o i tessuti sono per decenni stati appannaggio degli sviluppi accademici, anche per gli aspetti applicativi.
Le regole che governano la produzione di ATMPs e le difficoltà nella loro applicazione, specialmente in Europa
In Europa, la Commissione Europea ha prodotto negli ultimi anni una serie di normative per regolare la “vita” degli ATMPs, dalla scoperta alla (possibile) immissione nel mercato. La Direttiva più importante, in tal senso, è certamente la Direttiva Europea 2009/120/EC che si propone di regolare/controllare l’intero ciclo vitale di questi prodotti. Questa Direttiva, e molte altre successive, hanno di fatto equiparato questi prodotti ai normali “farmaci tradizionali” e richiedono, conseguentemente, che nella loro produzione siano pienamente rispettate le Buone Pratiche di Fabbricazione o GMP (Good Manufacturing Practices) che erano state, sin qui, di dominio esclusivo dell’Industria Farmaceutica.
Le Buone Pratiche di Fabbricazione o GMP (Good Manufacturing Practices) sono le linee guida che devono essere applicate alla fabbricazione di un qualsiasi farmaco, inclusi gli ATMPs, per assicurare che il farmaco somministrato sia conforme a requisiti minimi di qualità. Le GMP sono composte da diversi capitoli ed annessi che indicano con precisione tutti gli elementi di controllo richiesti durante la fabbricazione e sul prodotto stesso. Questi elementi includono i controlli sulle materie prime, i reattivi, eccipienti o altri additivi, i fornitori, i controlli di qualità e loro metodi, in itinere o sul prodotto finale, gli ambienti, i macchinari, il personale, i metodi di pulizia, la conservazione e quarantena dei prodotti e materiali, la gestione della qualità, ecc. Un elemento chiave delle GMP è la tracciabilità di tutti gli elementi che hanno concorso alla fabbricazione di un lotto specifico di medicinale. L’altro è il controllo del processo per garantirne la riproducibilità ed assicurare la sterilità del prodotto finale.
 Tuttavia i lotti prodotti generalmente o dedicati a pochi pazienti, e la natura biologica, e pertanto eterogenea e variabile, degli ATMPs fanno sì che la produzione di questi medicinali sperimentali non sempre si presta ad alcune delle regole di GMP pensate per i farmaci tradizionali. È da notare in modo particolare che gli ATMPs, essendo nella maggior parte dei casi composti di cellule vive, sono dei farmaci sterili e non sterilizzabili. Le GMP europee, a differenza di quelle americane, richiedono che tali farmaci sterili siano prodotti in ambienti di classe A (sterili) in un laboratorio in classe B (quasi sterile). Invece le norme GMP della Food and Drug Administration americana (FDA) richiedono la manipolazione di questi farmaci in classe A (sterile) in un laboratorio di classe C (con carica microbica e particellare meno stringente rispetto alla classe B). Questa apparente piccola differenza comporta una grande differenza nei costi e nella complessità dei laboratori di produzione. Infatti la classe B richiede molti più cambi d’aria nella stanza, un controllo particellare e microbiologico in continuo, una vestizione dell’operatore con tute speciali sterili, un maggior gradiente di pressione ecc. L’Europa è pertanto penalizzata rispetto agli USA nello sviluppo degli ATMPs, non solo a causa dei maggiori costi di produzione di questi farmaci, che derivano dalla legislazione europea significativamente più stringente di quella americana, ma anche a causa della maggior burocrazia e del minor finanziamento statale, almeno nella maggior parte degli Stati Europei. Questa “estremizzazione” è probabilmente figlia di una politica scelta dalla Commissione Europea “a monte” di tutta questa legislazione: l'“idea guida” è che prima “viene” la tutela della salute del consumatore/paziente e dunque le regole che garantiscono la riproducibilità, la tracciabilità, l'identificabilità univoca, la sterilità degli ambienti ecc., ovvero la sicurezza della non nocività che prevale sulla efficacia, la ricerca del rischio “0”. Tuttavia queste regole di produzione industriale hanno “violentato” la creatività pregressa degli stessi operatori/protagonisti: chi raccontasse come fu scoperto il trapianto di cellule staminali prima che vi fosse anche una sola vaga idea dei geni della compatibilità (HLA, Human Leukocyte Antigens, le proteine che identificano in modo univoco l'identità di ciascuno di noi), potrebbe concludere che con queste regole nuove avremmo fatto fatica oggi a “scoprirlo”. L’introduzione delle regole GMP ha generato una difficoltà anche concettuale in chi, per molti anni, aveva assistito al costante miglioramento “artigianale” del trapianto, che resta comunque una “terapia cellulare”, e che dunque mal comprendeva perché si dovesse passare a una logica industriale per queste altre terapie cellulari, spesso richieste/necessarie per problematiche cliniche insorte proprio durante un trapianto [tipico il caso delle preparazioni di sottopopolazioni di linfociti immuni del donatore (Donor Lymphocytes Infusions, DLI; Infusioni di Linfociti del Donatore) per trattare l’evento ricaduta/ripresa leucemica nel paziente trapiantato].
Tuttavia i lotti prodotti generalmente o dedicati a pochi pazienti, e la natura biologica, e pertanto eterogenea e variabile, degli ATMPs fanno sì che la produzione di questi medicinali sperimentali non sempre si presta ad alcune delle regole di GMP pensate per i farmaci tradizionali. È da notare in modo particolare che gli ATMPs, essendo nella maggior parte dei casi composti di cellule vive, sono dei farmaci sterili e non sterilizzabili. Le GMP europee, a differenza di quelle americane, richiedono che tali farmaci sterili siano prodotti in ambienti di classe A (sterili) in un laboratorio in classe B (quasi sterile). Invece le norme GMP della Food and Drug Administration americana (FDA) richiedono la manipolazione di questi farmaci in classe A (sterile) in un laboratorio di classe C (con carica microbica e particellare meno stringente rispetto alla classe B). Questa apparente piccola differenza comporta una grande differenza nei costi e nella complessità dei laboratori di produzione. Infatti la classe B richiede molti più cambi d’aria nella stanza, un controllo particellare e microbiologico in continuo, una vestizione dell’operatore con tute speciali sterili, un maggior gradiente di pressione ecc. L’Europa è pertanto penalizzata rispetto agli USA nello sviluppo degli ATMPs, non solo a causa dei maggiori costi di produzione di questi farmaci, che derivano dalla legislazione europea significativamente più stringente di quella americana, ma anche a causa della maggior burocrazia e del minor finanziamento statale, almeno nella maggior parte degli Stati Europei. Questa “estremizzazione” è probabilmente figlia di una politica scelta dalla Commissione Europea “a monte” di tutta questa legislazione: l'“idea guida” è che prima “viene” la tutela della salute del consumatore/paziente e dunque le regole che garantiscono la riproducibilità, la tracciabilità, l'identificabilità univoca, la sterilità degli ambienti ecc., ovvero la sicurezza della non nocività che prevale sulla efficacia, la ricerca del rischio “0”. Tuttavia queste regole di produzione industriale hanno “violentato” la creatività pregressa degli stessi operatori/protagonisti: chi raccontasse come fu scoperto il trapianto di cellule staminali prima che vi fosse anche una sola vaga idea dei geni della compatibilità (HLA, Human Leukocyte Antigens, le proteine che identificano in modo univoco l'identità di ciascuno di noi), potrebbe concludere che con queste regole nuove avremmo fatto fatica oggi a “scoprirlo”. L’introduzione delle regole GMP ha generato una difficoltà anche concettuale in chi, per molti anni, aveva assistito al costante miglioramento “artigianale” del trapianto, che resta comunque una “terapia cellulare”, e che dunque mal comprendeva perché si dovesse passare a una logica industriale per queste altre terapie cellulari, spesso richieste/necessarie per problematiche cliniche insorte proprio durante un trapianto [tipico il caso delle preparazioni di sottopopolazioni di linfociti immuni del donatore (Donor Lymphocytes Infusions, DLI; Infusioni di Linfociti del Donatore) per trattare l’evento ricaduta/ripresa leucemica nel paziente trapiantato].
I linfociti T possono essere ingegnerizzati mediante la introduzione stabile nel loro DNA di un gene “artificiale” prodotto in laboratorio, il così detto CAR. Questo gene codifica per una proteina di membrana, la cui porzione esterna alla cellule è un anticorpo in grado di riconoscere in modo selettivo un antigene bersaglio sulla superficie di cellule tumorali, e la porzione interna è molto simile al recettore tipico dei linfociti T (TCR, T Cell Receptor) e che, perciò, segnala attivamente all’interno delle cellule T attivandole. Il linfocita T, una volta ingegnerizzato, è in grado di riconoscere con alta specificità l’antigene tumorale e, quindi, di aderire al bersaglio (es. una cellula leucemia di tipo B, CD19 positiva) e di ucciderlo a seguito della attivazione. La rapidissima scoperta che i linfociti T armati dai CAR hanno una attività clinica molto importante (efficaci nel trattare fino al 90% dei casi di leucemica linfoblastica acuta di tipo B) ha scatenato una vera e propria corsa al coinvolgimento delle grandi multinazionali del farmaco (Pfizer, Glaxo Smith Kline, Cellgene, Novartis etc) che hanno prima sottoscritto accordi specifici con le realtà accademiche, tutte USA, che avevano sviluppato questa tecnologia in molti studi pilota di fase I/II (NIH, Baylor College Houston, US NCI, University Pennsylvania, Fred Hutchinson Cancer Center, Memorial Sloan Kettering e Seattle Children Research Center) e ora competono sul mercato globale per la diffusione di questi prodotti. Nessuna realtà accademica europea e nessuna realtà farmaceutica europea ha di fatto partecipato a questa competizione ed è possibile che in un futuro molto vicino questa opzione terapeutica sarà offerta a malati di leucemia/linfomi/mielomi di tipo B a costi elevatissimi e con una gestione molto complicata (saranno spese sostenibili per i sistemi sanitari nazionali europei?)
 A complicare ancora il quadro e rendere lo sviluppo degli ATMPs ancora più difficile è il fatto che l’industria non ha avuto, e probabilmente non ha ancora, grande interesse allo sviluppo di ATMPs di difficile e costosa produzione (lotti piccoli, dedicati a pochi pazienti o per malattie rare). Le uniche eccezioni possono essere alcuni prodotti che sembrano mostrare molta efficacia ed essere applicabili a grandi numeri di pazienti, come nel caso dei linfociti T modificati geneticamente con il Chimeric Antigen Receptor (CAR-T, potenzialmente applicabili a molti tumori). Ma anche in questi casi, l’industria tende ad entrare in campo solo dalla fase III dello sviluppo del farmaco in poi, e lascia che l’accademia faccia gli studi preclinici e clinici di fase I/II. Quindi l'identificazione delle potenziali “scoperte” e l’ottimizzazione delle idee scientifiche nella loro finalizzazione terapeutica è di fatto un'area lasciata all'accademia e alle fasi I/II sperimentali, e la rigidità della applicazione delle GMP in Europa rischia di essere letale su questa fase di necessità vivace e massimamente esplorativa. È molto importante sottolineare qui che tutti i risultati scientifici/clinici delle realtà accademiche sopra riportate, sono stati prodotti con produzioni “in house” nella realtà americana, controllata e regolata dalla FDA, con i criteri più blandi di quelli equivalenti definiti dalla Unione Europea. Paradossalmente, poi, la rigidità della normativa finisce per compromettere anche l'industria stessa nelle fasi di sviluppo successive: i 318 diversi trials clinici sperimentali con 250 ATMPs registrati nel database ufficiale [European Union Drug Regulating Authorities Clinical Trials (EudraCT)] dal 2004 al 2012 erano per il 60% supportati da organizzazioni accademiche o Charities. Come riportato di recente dagli stessi membri del Comitato per le Terapie Avanzate dell’EMA (Committee for Advanced Therapies - CAT), solo un prodotto di terapia genica (Glybera) e 4 prodotti di terapia cellulare (ChondroCelect, MACI, Provenge e Holoclar) hanno “raggiunto" la MA1 . Inoltre, altri due prodotti di terapia genica potrebbero essere presto commercializzati (Imlygic e il gene ADA). È interessante osservare che il MACI è stato ritirato dal commercio e il Provenge ha ottenuto risultati negativi in fase III tali da portare al fallimento della ditta produttrice.
A complicare ancora il quadro e rendere lo sviluppo degli ATMPs ancora più difficile è il fatto che l’industria non ha avuto, e probabilmente non ha ancora, grande interesse allo sviluppo di ATMPs di difficile e costosa produzione (lotti piccoli, dedicati a pochi pazienti o per malattie rare). Le uniche eccezioni possono essere alcuni prodotti che sembrano mostrare molta efficacia ed essere applicabili a grandi numeri di pazienti, come nel caso dei linfociti T modificati geneticamente con il Chimeric Antigen Receptor (CAR-T, potenzialmente applicabili a molti tumori). Ma anche in questi casi, l’industria tende ad entrare in campo solo dalla fase III dello sviluppo del farmaco in poi, e lascia che l’accademia faccia gli studi preclinici e clinici di fase I/II. Quindi l'identificazione delle potenziali “scoperte” e l’ottimizzazione delle idee scientifiche nella loro finalizzazione terapeutica è di fatto un'area lasciata all'accademia e alle fasi I/II sperimentali, e la rigidità della applicazione delle GMP in Europa rischia di essere letale su questa fase di necessità vivace e massimamente esplorativa. È molto importante sottolineare qui che tutti i risultati scientifici/clinici delle realtà accademiche sopra riportate, sono stati prodotti con produzioni “in house” nella realtà americana, controllata e regolata dalla FDA, con i criteri più blandi di quelli equivalenti definiti dalla Unione Europea. Paradossalmente, poi, la rigidità della normativa finisce per compromettere anche l'industria stessa nelle fasi di sviluppo successive: i 318 diversi trials clinici sperimentali con 250 ATMPs registrati nel database ufficiale [European Union Drug Regulating Authorities Clinical Trials (EudraCT)] dal 2004 al 2012 erano per il 60% supportati da organizzazioni accademiche o Charities. Come riportato di recente dagli stessi membri del Comitato per le Terapie Avanzate dell’EMA (Committee for Advanced Therapies - CAT), solo un prodotto di terapia genica (Glybera) e 4 prodotti di terapia cellulare (ChondroCelect, MACI, Provenge e Holoclar) hanno “raggiunto" la MA1 . Inoltre, altri due prodotti di terapia genica potrebbero essere presto commercializzati (Imlygic e il gene ADA). È interessante osservare che il MACI è stato ritirato dal commercio e il Provenge ha ottenuto risultati negativi in fase III tali da portare al fallimento della ditta produttrice.
Il futuro e le modalità per superare le difficoltà
Queste storie di ricerca, non esaustive della situazione, sottolineano da un lato l’entusiasmante “verifica” nel terreno della malattia di quanto per decenni era stato “scoperto” (dai farmaci “specifici” per le lesioni genetiche puntiformi dei tumori, all'identificazione di tutte le molecole coinvolte nel contatto tra cellule del sistema immunitario e cellule tumorali, con successiva identificazione di molecole “specifiche” per bloccare/sbloccare alcune tra queste interazioni), ma, d’altro canto, pongono la questione che la “traducibilità” della ricerca, dal bancone al letto dell’ammalato (abusatissima espressione), richiede un impegno “industriale” o l’affidamento tout court alla industria tradizionale per la loro gestione.
Questo è lo scenario per chi, come noi, aveva per molti anni “sognato” che le conoscenze si potessero tradurre in salute e oggi verifica (ed è una immensa felicità) che molte cure si affacciano “sfruttando” quei dati, ma si deprime nella constatazione di dover “passare” la mano ad altri per l’operatività (e non più con l’originale pensiero della gratuità e della idealità, ma con regole diverse).
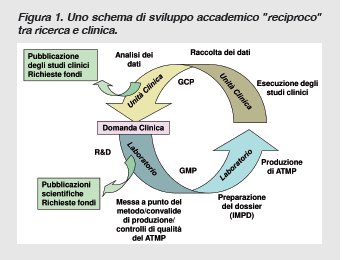
Da tutto questo discende la nostra decisione, nel 2003, di provare a costruire una Cell Factory a “norma” secondo le GMP industriali, all’interno dell’Ospedale Pubblico (prima Riuniti, ora Papa Giovanni XXIII), farsela regolarmente autorizzare da AIFA (Agenzia Italiana del Farmaco), gestirla con medici/biologi/ricercatori su fondi di ricerca o di Charities no profit private e, infine, disegnare studi clinici di fase I/II non sponsorizzati, farli autorizzare dalle Autorità Competenti e condurli, comunque secondo le regole delle GCP, in piena autonomia e libertà nel nostro Ospedale.
In questo modo volevamo salvaguardare il flusso della vita scientifica nella nostra realtà, che inizia a partire da una domanda clinica, si snoda attraverso attività laboratoristiche di ricerca e sviluppo al bancone di laboratorio, si focalizza su dossier specifici da sottoporre alle autorità competenti quando un prodotto sembra ipotizzabile per uno sviluppo clinico (chiamati IMPD Investigational Medical Product Dossier, contenenti anche il protocollo clinico vero e proprio) e quindi torna alla clinica dove l’ipotesi viene testata. Il tutto con la democratica e verificabile attitudine a pubblicare sia i risultati della ricerca preclinica che i risultati della ricerca clinica. La nostra Cell Factory, costruita nel 2002 con l’aiuto finanziario, e non solo, della Associazione Italiana Lotta alla Leucemia (AIL) Paolo Belli di Bergamo, è composta fondamentalmente da due stanze in classe B e due stanze in classe C (sono definizioni di zone a contaminazione controllata, anche note come camere bianche). È stata autorizzata per la prima volta nel 2007 e da allora riceve ogni due anni successive autorizzazioni a seguito di intense visite ispettive da parte di AIFA. Un modo per abbattere i costi di questa impresa senza capitale e senza vendita è stato quello di utilizzare lo stesso personale (per lo più contrattisti e borsisti) che opera nella struttura anche per la “normale” manipolazione delle cellule ematopoietiche staminali per il trapianto e che richiede, anche questa parte, l’adeguamento ad altre complesse normative secondo standard internazionali. Perciò l’ottimizzazione del personale, strutture, macchinari, materiali e attività di assicurazione della qualità hanno consentito di rendere fin qui l’impresa sostenibile. Che questo tipo di impresa sia difficile lo dicono anche i numeri: • se si guarda il registro “ufficiale” delle terapie cellulari e geniche a cura dell’Istituto Superiore di Sanità (ISS) (in cui si sono registrati quelli che hanno “voluto” registrarsi, in questa “area” il “nero” di chi opera senza dichiararsi è illimitato, vedi vicenda Stamina Foundation2,3 e tutto il mondo “in rete” che offre miracolose terapie a base di cellule staminali) si osserva che, dal 2005 ad oggi, sono stati registrati 51 studi clinici (12 con terapia genica e 39 con terapia cellulare somatica) che risultano “chiusi” e 19 studi clinici (6 con terapia genica e 13 con terapia cellulare somatica) che risultano “aperti”; • gli studi “chiusi” dovrebbero essere stati pubblicati, seguendo la logica di una etica della ricerca scientifica e, solo così, potrebbe essere verificata a posteriori la loro qualità ed efficacia4-9 ; • se si guarda agli studi aperti è interessante osservare che le realtà attive oggi per la terapia cellulare somatica sono 7: Bergamo (7 studi), Monza S. Gerardo (2 studi), Policlinico Milano (2 studi), FabioCell Roma (1 studio), Ospedale di Terni (1 studio), Istituto tumori di Meldola (1 studio), H S Raffaele Milano (1 studio) e solo 1 attiva per le terapie geniche, H S Raffaele Milano (4 studi).
Nella tabella 1 riportiamo gli studi in corso, la numerosità dei pazienti coinvolti ad oggi (11/11/2014), la patologia, il tipo cellulare utilizzato e la sede della Cell Factory coinvolta nel processo produttivo. 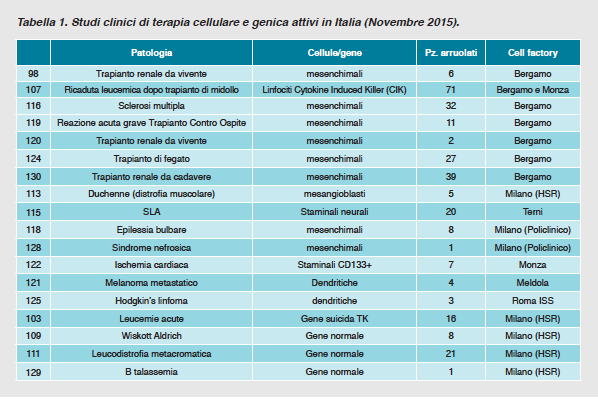 Non si può non rilevare che, nel corso degli ultimi anni, il numero già ridotto delle Cell Factories italiane si è ulteriormente ridotto a causa di problemi di in-sostenibilità economica o per non corrispondenza con gli standards operativi/qualitativi richiesti (8 realtà attive delle originali 13 strutture costruite, 11 hanno mantenuto l'autorizzazione). Per contro, alla luce di quanto visto già con alcune terapie geniche (cura di alcune immunodeficienze congenite) e con alcune terapie cellulari di successo (cellule staminali della cornea) è auspicabile che questa rete di Cell Factories possa estendersi e possa sostenersi con finanziamenti istituzionali in appoggio del tradizionale sostentamento da parte dei fondi di ricerca e delle Charities.
Non si può non rilevare che, nel corso degli ultimi anni, il numero già ridotto delle Cell Factories italiane si è ulteriormente ridotto a causa di problemi di in-sostenibilità economica o per non corrispondenza con gli standards operativi/qualitativi richiesti (8 realtà attive delle originali 13 strutture costruite, 11 hanno mantenuto l'autorizzazione). Per contro, alla luce di quanto visto già con alcune terapie geniche (cura di alcune immunodeficienze congenite) e con alcune terapie cellulari di successo (cellule staminali della cornea) è auspicabile che questa rete di Cell Factories possa estendersi e possa sostenersi con finanziamenti istituzionali in appoggio del tradizionale sostentamento da parte dei fondi di ricerca e delle Charities.
Molte sperimentazioni cliniche in corso, come si evince dalla tabella, sono a base di cellule “mesenchimali”. Queste cellule, di fatto, costituiscono lo stroma di diversi organi, in particolare del midollo osseo o della parete del cordone ombelicale e possono essere cresciute con semplicità in laboratorio fino a raggiungere grandi numeri. La loro natura parzialmente indifferenziata, è tale per cui possono, se sottoposte a stimoli chimici, differenziarsi verso cellule ossee, o della cartilagine o cellule adipose o altro ancora. Tuttavia la loro più interessante caratteristica è quella di esercitare in vivo un effetto immunomodulante, per lo più antinfiammatorio e immuno-soppressorio, probabilmente a causa della loro rapida localizzazione nelle sedi in cui avviene l’incontro tra l’antigene e il sistema immunitario dell’ospite (per esempio i linfonodi) e del rilascio di grandi quantità di molecole solubili che condizionano e orchestrano lo stato funzionale del tessuto in senso tollerogenico. Questo giustifica il loro utilizzo in tanti programmi clinici in cui si vuole cercare di conferire una linea di immunosoppressione aggiuntiva a quelle farmacologiche tradizionali (es nella GVHD, nel trapianto di rene e di fegato, nella sclerosi multipla ecc.).
Conclusioni 
Il modello del Centro di Terapia Cellulare “G.Lanzani” di Bergamo, che abbiamo presentato, ci ha consentito di fare della ricerca innovativa nel campo delle ATMPs, ma anche di produrre studi clinici sperimentali di fase I/II. Tutto questo è stato ottenuto senza alcuna ambizione di voler “raggiungere il mercato” (Market Authorisation) ma, piuttosto, con l’obiettivo di identificare nuove opzioni terapeutiche per migliorare condizioni critiche in un gruppo relativamente piccolo di pazienti che fanno parte della nostra pratica clinica quotidiana. Come detto sopra, gli ATMPs rappresentano dei tipici prodotti di “ricerca” in evoluzione e il campo è stato sviluppato da comunità di ricercatori di base e clinici, che includono le più avanzate competenze, in contesti laboratoristici/sperimentali, non confrontabili con le classiche situazioni non di sviluppo-produzione industriale, che obbediscono a criteri fortemente rigidi, quali quelli necessari per la commercializzazione su larga scala. L’applicazione rigida a scenari in cui la componente di ricerca-sperimentazione è centrale di aspetti soprattutto procedurali della legislazione del farmaceutico industriale può essere di fatto uno dei fattori di rischio più grandi (ed evitabili) per la vitalità del settore. È chiaro che gli standard di qualità e sicurezza sono imprescindibili. È altrettanto auspicabile (come ulteriore passo in avanti, al di là delle tappe, non facili, lungo le quali si è costruita una relazione di reciproco rispetto tra chi ricerca-produce e chi controlla) una applicazione intelligente delle norme. Il termine più ovvio e diretto di riferimento (anche in coerenza con le tendenze generali a processi di armonizzazione “globale” del farmaceutico) è quanto ha caratterizzato (non sempre ovviamente in modo esemplare) la legislazione degli USA. Da una parte per una gestione efficiente degli studi accademici di fase I/II, dall’altra con la creazione di reti funzionali tra cell factories certificate, formalmente riconosciute come risorsa strategica di salute pubblica, ed in quanto tale sostenute da fondi istituzionali, che possano garantire autonomia, sostenibilità, competitività: con investimenti per cui siano chiari e non punitivi i criteri di valutazione e rendicontazione integrata, scientifica ed economica.