Medicinale soggetto a prescrizione medica limitativa, vendibile al pubblico su prescrizione di centri ospedalieri o di specialisti: cardiologo, pneumologo, reumatologo (RRL).
Prescrizione soggetta a diagnosi - piano terapeutico - PHT
Medicinale sottoposto a monitoraggio addizionale.
Classe A Indicazioni registrate: trattamento a lungo termine dell'ipertensione arteriosa polmonare (IAP) in pazienti adulti in Classe Funzionale (FC) WHO II e III, sia come terapia di combinazione nei pazienti controllati in maniera insufficiente con un antagonista recettoriale dell’endotelina (ERA) e/o un inibitore della fosfodiesterasi di tipo 5 (PDE-5), sia in monoterapia nei pazienti che non sono candidabili a tali terapie.
Proprietà farmacologiche
Selexipag è un agonista orale selettivo dei recettori prostanoidi IP, approvato prima dalla FDA e poi autorizzato anche da EMA il 12/05/2016 (in Italia è disponibile dal 22/01/2018) per il trattamento dell’ipertensione arteriosa polmonare (IAP).
Il selexipag e il suo potente metabolita attivo (37 volte più potente del selexipag) hanno un meccanismo d’azione simile a quello della prostaciclina endogena: sono agonisti del recettore IP, ma ad alta affinità e con un'elevata selettività per questo recettore rispetto ai recettori prostanoidi EP1–EP4, DP, FP e TP. I recettori EP1, EP3, FP e TP sono recettori contrattili ben descritti nell'apparato gastrointestinale e nei vasi sanguigni; i recettori EP2, EP4, e DP1 mediano gli effetti immunodepressivi. La stimolazione del recettore IP porta ad effetti vasodilatatori, anti-proliferativi e anti-fibrotici.
Negli studi preclinici su animali selexipag ha dimostrato di prevenire il rimodellamento cardiaco e polmonare e provocare una diminuzione proporzionale della pressione polmonare e periferica, indicando che la vasodilatazione periferica riflette l'efficacia farmacodinamica a livello polmonare1. La selettività di selexipag e del suo metabolita attivo per i recettori IP fa sì che, almeno in teoria, potrebbero causare meno effetti avversi gastrointestinali (nausea e vomito)2.
Dopo una singola dose, le concentrazioni plasmatiche massime (Cmax) di selexipag e del suo metabolita attivo vengono raggiunte rispettivamente in 1-3 e 3-4 ore. L’assunzione di selexipag con il cibo può migliorarne la tollerabilità gastrointestinale; più soggetti hanno riportato eventi avversi dopo la somministrazione a stomaco vuoto rispetto a stomaco pieno. Il selexipag viene metabolizzato principalmente a livello epatico: prima in metabolita attivo tramite idrolisi, poi in metaboliti inattivi per via epatica tramite CYP3A4 e 2C8 e glucuronidazione mediante UGT1A3 e 2B7. L’eliminazione avviene prevalentemente nelle feci (93%). L’emivita di eliminazione terminale media è 0,8-2,5 ore per il selexipag e 6,2-13,5 ore per il suo metabolita attivo1.
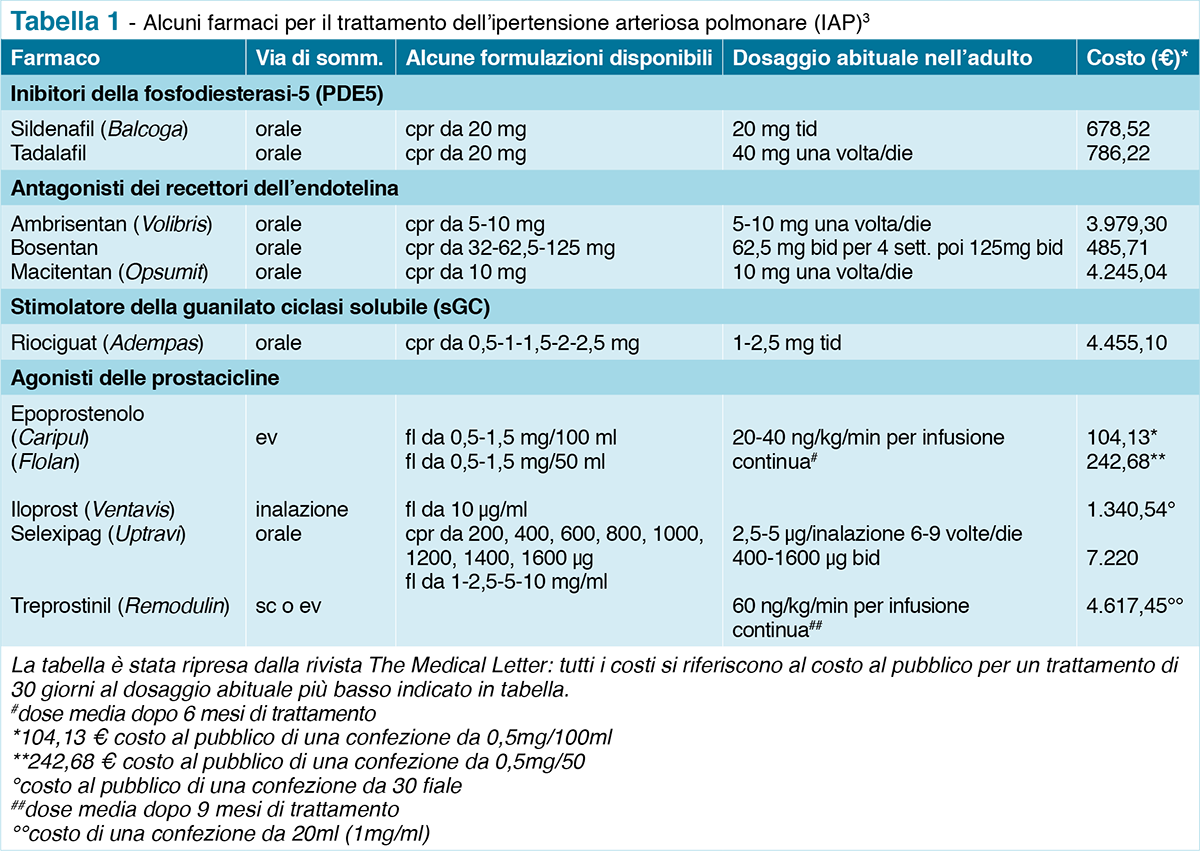
Rispetto ad altri medicinali della classe degli agonisti delle prostacicline (epoprostenolo, iloprost, treprostinil) che vengono somministrati in vena, Selexipag ha il vantaggio di poter essere assunto per via orale3.
Efficacia clinica
L’ipertensione arteriosa polmonare è caratterizzata da un progressivo aumento delle resistenze vascolari polmonari che portano ad una insufficienza del ventricolo destro con sintomi correlati quali dispnea a riposo (classe IV NYHA/OMS) o dopo sforzi minimi (Classe III), e ad una mortalità precoce. I sintomi sono affanno, vertigini e stanchezza.
Per il trattamento iniziale della IAP (classe funzionale OMS I e II) si raccomanda in genere la monoterapia con un farmaco orale. I farmaci approvati dalla FDA per questa indicazione comprendono: gli inibitori della fosfodiesterasi-5 (PDE5) (sildenafil, tadalafil), gli antagonisti dei recettori dell’endotelina (ambrisentan, bosentan, macitentan) e uno stimolante della guanilato ciclasi (riociguat). I pazienti con malattia più avanzata possono essere trattati con prostaciclina (epoprostenolo, iloprost, treprostinil) (Tabella 1).
Le attuali linee guida statunitensi raccomandano il trattamento con due o più classi di farmaci solo quando la risposta è inadeguata o il paziente peggiora durante la monoterapia4. Le linee guida europee, di più recente pubblicazione, includono raccomandazioni per un’iniziale terapia di associazione5.
L’approvazione del selexipag si è basata sui risultati di uno studio di fase 3, multicentrico, in doppio cieco, randomizzato e controllato con placebo (GRIPHON) condotto su 1.156 pazienti con IAP sintomatica (classe funzionale OMS II o III), naive al trattamento o già in trattamento con un antagonista dei recettori dell’endotelina e/o con un inibitore della fosfodiesterasi-5. L’endpoint primario era il tempo alla comparsa entro la fine del trattamento del primo evento di morbilità o mortalità. Si trattava di un endpoint composito che includeva: morte per qualsiasi causa, ospedalizzazione per IAP, o progressione della IAP con conseguente necessità di trapianto polmonare o settostomia atriale con palloncino, o inizio di una terapia parenterale a base di prostanoidi o di un'ossigenoterapia cronica, o altri eventi legati alla progressione della malattia confermata da una diminuzione rispetto al basale (≥15%) della distanza percorsa in 6 minuti (6MWD, 6-minute walk distance) e un peggioramento della classe funzionale OMS (per i pazienti in OMS FC II o III al basale) o confermata da una diminuzione della 6MWD rispetto al basale (≥15%) e la necessità di una terapia aggiuntiva specifica per la IAP (per i pazienti in OMS FC III o IV al basale). La durata mediana del trattamento era di 70,7 settimane con il selexipag e di 63,7 settimane con il placebo.
I risultati dello studio hanno mostrato che il trattamento con selexipag (titolato alla massima dose tollerata fino a 1600 µg due volte al giorno) era capace di ridurre del 40% rispetto al placebo (hazard ratio 0,60; IC 99% (0,46; 0,78); p< 0,0001) l’insorgenza di eventi di morbi-mortalità6.
L’aumento del numero di decessi, riscontrato fino alla fine del trattamento + 7 giorni nel gruppo selexipag rispetto al gruppo placebo (rispettivamente 46/574 corrispondenti all’8,0% vs. 37/582 corrispondenti al 6,4%), ma non osservato alla chiusura dello studio (17,4%; 100/574 nel gruppo selexipag vs. 18,0%; 105/582 nel gruppo placebo), è stato ulteriormente valutato attraverso modelli matematici che hanno dimostrato che lo sbilanciamento dei decessi è consistente con l’ipotesi di un effetto neutrale sulla mortalità per IAP e la riduzione degli eventi non fatali7 (non hanno quindi mostrato una riduzione della mortalità nel gruppo trattato con selexipag rispetto al gruppo placebo). Tuttavia tali risultati hanno aperto una discussione sull’impiego del selexipag nella pratica clinica: per esempio nel 2016 la commissione farmaco-economica francese ha concluso che selexipag non forniva un beneficio clinico sufficiente nei pazienti con IAP per essere rimborsabile8, mentre il CHMP dell’EMA li ha ritenuti ininfluenti e ne ha pertanto autorizzato l’immissione in commercio7.
La distanza percorsa nel test del cammino dei 6 minuti a 26 settimane, un endpoint secondario, è aumentata in maniera significativa rispetto al basale con il selexipag (+9,0 vs. -4,0 metri con il placebo)6.
Per avere dati più consistenti si dovrà attendere i risultati dei due studi post-autorizzazione proposti dal CHMP dell’EMA attraverso il piano di gestione del rischio. Si tratta di uno studio osservazionale di coorte sui pazienti con IAP esposti o meno a selexipag nella pratica clinica con lo scopo di: descrivere le caratteristiche epidemiologiche e cliniche dei pazienti, complessivamente e nel sottogruppo di pazienti con età > 75 anni; caratterizzare ulteriormente il profilo di sicurezza del farmaco e stimare l’incidenza di morte per qualsiasi causa e dei rischi identificati come importanti (ipotensione, anemia) o come potenzialmente importanti (edema polmonare associato con malattia veno-occlusiva, ipertiroidismo, eventi cardiovascolari maggiori, insufficienza renale, sanguinamento, …); confrontare il tasso di eventi cardiovascolari maggiori e di morte per qualsiasi causa tra i pazienti esposti o meno a selexipag. I risultati sono previsti per il 2023.
Il secondo studio ha lo scopo per valutare le misure di minimizzazione del rischio di errore medico durante la fase di titolazione e registrare la frequenza di dosi sbagliate riportate dal paziente stesso. Lo studio dovrebbe terminare nel 20207.
Effetti indesiderati1
Le reazioni avverse più comunemente riportate sono cefalea, diarrea, nausea e vomito, dolore alla mandibola, mialgia, dolore alle estremità, artralgia e sintomi simil-influenzali. Queste reazioni sono più frequenti durante la fase di titolazione. La maggior parte di queste reazioni è di lieve o moderata intensità.
Selexipag ha proprietà vasodilatatorie sull’intero sistema vascolare che potrebbero comportare anche rossore con vampate di calore, seri effetti avversi cardiovascolari come ipotensione e attacchi di angina in pazienti con malattia coronarica.
Avvertenze1
Non è necessario alcun aggiustamento del regime di dose nei pazienti anziani (≥65 anni). L'esperienza clinica in pazienti di oltre 75 anni è limitata, pertanto selexipag deve essere utilizzato con cautela in questa popolazione.
Il farmaco non deve essere somministrato nei pazienti con compromissione epatica severa (Classe C Child-Pugh). In quelli con compromissione epatica moderata (Classe B Child-Pugh), la titolazione deve essere eseguita con cautela. Quelli con compromissione epatica lieve (Classe A Child-Pugh) non richiedono alcun aggiustamento del regime di dose.
Nei pazienti con compromissione renale lieve o moderata non è necessario alcun aggiustamento del regime di dose, mentre in quelli con compromissione renale severa la titolazione deve essere eseguita con cautela.
La sicurezza e l'efficacia del selexipag nei soggetti di età compresa fra 0 e 18 anni non è ancora stata stabilita e non vi sono dati disponibili. Pertanto la somministrazione di selexipag in questa popolazione non è raccomandata.
Dosaggio
Aumento graduale della dose (titolazione) fino alla dose massima tollerata personalizzata, che può essere compresa tra 200 e 1600 µg, somministrata due volte al giorno.
Costi
Il costo al pubblico per un trattamento di 30 giorni con selexipag (Uptravi®), somministrato due volte/die, è di € 7.2209, costo più alto sia rispetto agli altri agonisti delle prostacicline sia rispetto agli altri farmaci disponibili per il trattamento dell’IAP (Tabella 1)3.
Bibliografia 1. UPTRAVI. Riassunto delle caratteristiche del prodotto. Disponibile sul sito http://www.ema.europa.eu (ultimo accesso 02/06/2018). 2. Morrison K, et al. Selexipag: a selective prostacyclin receptor agonist that does not affect rat gastric function. J Pharmacol Exp Ther 2010;335:249-55. 3. Selexipag (Uptravi) for Pulmonary Arterial Hypertension. The Medical Letter on Drugs and Therapeutics, Anno XLV, n. 8, 15 aprile 2016, pag. 45. 4. Taichman DB, et al. Pharmacologic therapy for pulmonary arterial hypertension in adults: CHEST guideline and expert panel report. Chest 2014; 146:449-475. 5. Galiè N, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J 2016; 37:67–119. 6. Sitbon O, et al. Selexipag for the Treatment of Pulmonary Arterial Hypertension. N Engl J Med 2015; 373:2522-33. 7. EMA, Committee for Medicinal Products for Human Use (CHMP), 2016. Assessment report: Uptravi. Procedure No. EMEA/H/C/003774/0000, 01 April 2016. EMA/272184/2016. Disponibile sul sito http://www.ema.europa.eu (ultimo accesso 02/06/2018). 8. Selexipag and pulmonary arterial hypertension. Prescrire International 2017; 26: 237-8. 9. AIFA. DETERMINA 20 dicembre 2017. Classificazione del medicinale per uso umano «Uptravi» ai sensi dell’art. 8, comma 10, della legge 24 dicembre 1993, n. 537. (Determina n. 2105/2017). (GU Serie Generale n.16 del 20-01-2018).
Selexipag è un agonista della prostaciclina. Rispetto agli altri agonisti della prostaciclina disponibili per il trattamento della IAP, è selettivo nei confronti dei recettori prostanoidi IP e può essere somministrato per via orale.
Le evidenze disponibili si basano sui risultati di uno studio clinico randomizzato controllato vs. placebo: selexipag riduce il rischio di progressione della malattia e di ospedalizzazione nei pazienti con ipertensione arteriosa polmonare (IAP), ma non è stato dimostrato che riduca la mortalità. Non è stato confrontato con nessuno degli altri farmaci della classe degli agonisti delle prostacicline (non disponibili per via orale) o con altri farmaci orali che hanno questa stessa indicazione terapeutica. Inoltre, i seri effetti indesiderati cardiovascolari e i dubbi riguardo l’aumento del numero di decessi fra i pazienti trattati col farmaco, ne sconsigliano per ora l’utilizzo.