Paolo Furno, Gianpaolo Bucaneve, Albano Del Favero Sezione di Medicina Interna e Scienze Oncologiche, Policlinico Monteluce, Perugia
Novità terapeutiche
Una presentazione dell'articolo che segue è opportuna per tre obiettivi complementari:
richiamare la attualità e la centralità delle tematiche cui è stato dedicato il n. 1 2002 di questa rivista. Averle commentate come "evento sentinella" non avrebbe senso, se non diventano uno strumento normale di lettura e di interpretazione delle sezioni specifiche (v. Bussola e non solo) che via via presentano le novità che il mercato propone.
Concentrare l'attenzione (di contenuti e di metodo) in questo numero della rivista su due casi concreti che sono ben esemplificativi e didattici sul che cosa significa prendere sul serio una valutazione complessiva della innovatività terapeutica:
a. nella Bussola occupano uno spazio importante schede informative relative a situazioni cliniche che rientrano nella definizione di "malattie rare". Non è difficile riconoscere nei profili proposti elementi di perplessità importanti sulla qualità dei dati che stanno alla base delle registrazioni dei farmaci di interesse. C'è una contraddizione evidente tra le richieste (giustificatissime!) di dare priorità a problemi seri che non hanno ancora risposte terapeutiche soddisfacenti, e la povertà metodologica con la quale la serietà delle condizioni cliniche è affrontata nella sperimentazione. Alla serietà di domande-bisogni sembra corrispondere una proporzionale non-serietà dei criteri con cui si cercano le risposte. La serietà sembra non evocare una coerenza, ma una dispensa dal rigore di ricerca.
b. La rassegna che segue è dedicata alle "novità" nel campo degli antibiotici. La forma, la lunghezza, il dettaglio di quanto è proposto sono palesemente diversi da quelli di solito adottati (e questa è solo la prima parte di un lavoro, che avrà una seconda puntata). Si è pensato potesse essere utile, per una volta, far seguire nel dettaglio tutta l'articolazione di un percorso informativo, di solito compattato in 1-2 pagine. La scelta degli antibiotici è particolarmente utile per dare un'idea concreta delle tante variabili (conoscitive e assistenziali) che devono entrare nel processo valutativo.
Collegare, ricondurre, integrare le tematiche delle innovazioni/avanzamenti terapeutici al percorso di riflessione e proposte dell'Editoriale sulla psicofarmacologia. Il richiamo non è nuovo, ma (vista la realtà) val sempre la pena ripeterlo: i farmaci non sono mai valutabili "in isolamento", non importa quanto il loro uso possa apparire determinato da criteri "oggettivi". Ciò che appassiona del lavoro di informare/si è proprio questo esercizio permanente di essere rimandati dai farmaci ai problemi-bisogni per i quali sono indicati.
Diversi nuovi farmaci antibatterici sono entrati in commercio nell'ultimo anno ed alcuni altri sono in procinto di farlo. Il continuo proliferare di nuovi antibiotici insieme ad una promozione "marketing" aggressiva possono indurre nel medico un certo disorientamento prescrittivo, riguardo al come e quando utilizzare ciò che è già da tempo a disposizione nell'armamentario terapeutico oppure ricorrere a ciò che viene presentato come innovativo e potenzialmente vantaggioso.
L'innovatività del più recente antibiotico che viene ad aggiungersi ai precedenti dovrebbe essere valutata sulla base di precisi criteri di giudizio al fine di discriminare tra una sostanziale novità o, invece, una "pseudo-novità". Tali criteri riguardano:
1. le reali necessità terapeutiche anche emergenti, ovvero ciò di cui abbiamo, o potremo avere a breve, più impellente bisogno in antibiotico-terapia; 2. le caratteristiche del farmaco in questione, ovvero ciò che il farmaco offre; 3. le alternative terapeutiche in uso, ovvero il confronto clinico comparativo con i farmaci di cui già disponiamo (tabella 1).
Senza dubbio una delle principali necessità terapeutiche emergenti negli ultimi anni nel campo delle infezioni batteriche, che stimolano lo sviluppo di molecole con attività antimicrobica innovativa, è rappresentata dal problema delle antibiotico-resistenze multiple, ovvero dalla insorgenza e dalla crescente e rapida disseminazione su scala globale di patogeni che hanno acquisito resistenza verso molteplici farmaci antibatterici da tempo in uso, capaci di sostenere infezioni comuni e gravi, potenzialmente fatali 1-3 (tabella 2).
Questa inesorabile tendenza, che pur presenta ancora vistose differenze regionali nelle proporzioni epidemiologiche del problema, ha avuto paradossalmente come concausa maggiore proprio la crescente, massiccia e spesso indiscriminata utilizzazione di antibiotici verificatasi soprattutto negli ultimi due decenni soprattutto in relazione all'aumento generale della popolazione dei pazienti immunocompromessi ad alto rischio infettivo (oncologici, neutropenici, trapiantati, HIV-positivi, in terapia immunosoppressiva, ecc.).
Se da un lato quindi l'emergente problema dei patogeni multiresistenti crea senza dubbio la reale necessità clinico-terapeutica di sviluppare rapidamente farmaci antibatterici nuovi, che siano attivi soprattutto su quei patogeni contro i quali le opzioni terapeutiche a disposizione al momento si vanno progressivamente erodendo, dall'altro esso viene ancora più a sottolineare la vitale necessità di un uso sempre più appropriato e controllato dei farmaci antibatterici già esistenti, allo scopo di salvaguardarne la validità clinica presente e futura il più a lungo possibile. Quest'ultima necessità è quanto mai importante per i farmaci antibatterici di più recente introduzione, al fine di evitare di alimentare ulteriormente l'emergere di antibiotico-resistenze e la conseguente paradossale rincorsa verso il "nuovo" antibiotico.
I farmaci antibatterici di recente introduzione che saranno presi in considerazione in questo articolo e nel successivo (quinupristin-dalfopristin, linezolid, telitromicina, gatifloxacina, moxifloxacina, ertapenem) in generale sembrano offrire un'attività antimicrobica più accentuata ed ampia (che comprende in particolare i patogeni antibiotico-resistenti) rispetto ai farmaci già a disposizione. Si cercherà qui di esaminarne sinteticamente le caratteristiche potenzialmente vantaggiose e svantaggiose, di capire se siano realmente migliori dei vecchi farmaci, ovvero se, ed in che misura, essi rappresentino un sostanziale avanzamento in antibiotico-terapia nel senso di una comprovata superiorità nel trattamento delle infezioni sostenute dai patogeni multiresistenti o dai patogeni più comuni e, conseguentemente, di delinearne il possibile attuale ruolo in antibiotico-terapia rispetto all'armamentario antibatterico già esistente.
Struttura e attività
Quinupristin/dalfopristin (Synercid, Aventis Pharma, flac. e.v. 500 mg 10 ml, € 89,10 ) è il primo agente antibatterico appartenente alla classe delle streptogramine per uso sistemico parenterale. Ciascuno dei due composti, derivati semisintetici della pristinamicina, possiede una certa attività antibatterica, ma questa risulta drasticamente aumentata per sinergismo nella combinazione in rapporto ponderale 30:70. Lo spettro di attività antibatterica è piuttosto selettivo e comprende segnatamente quasi tutti i Gram-positivi, inclusi i ceppi resistenti alla eritromicina, penicillina, oxacillina ed ai glicopeptidi, quali: stafilococchi, E. faecium, S. pneumoniae, S. pyogenes, streptococchi viridanti, Peptostreptococcus spp e Clostridium perfringens. Meno costante è l'attività su Listeria monocytogenes, Corynebacterium JK ed altri anaerobi Gram-positivi; il farmaco non è attivo su E. faecalis. Quinupristin/dalfopristin inibisce anche alcuni patogeni Gram-negativi come Moraxella catarrhalis, Legionella pneumophila, Mycoplasma pneumoniae, Neisseria meningitidis, ma è inattivo su Haemophilus influenzae, enterobacteriacee e P. aeruginosa. In vitro il farmaco è battericida verso molti ceppi di stafilococco e streptococco, ma è solo batteriostatico contro E. faecium; un effetto post-antibiotico è dimostrabile in vitro nei confronti dei Gram-positivi. Rifampicina e doxiciclina sono antibatterici sinergici con quinupristin/dalfopristin rispettivamente contro stafilococco aureo meticillino-resistente (MRSA) ed E. faecium vancomicino-resistente (VREF); contro quest'ultimo anche vancomicina ed ampicillina mostrano un certo grado di sinergismo in vitro con quinupristin/dalfopristin. Negli studi clinici con il farmaco, l'insorgenza di resistenza in corso di trattamento è stata documentata in E. faecium con frequenza variabile dal 4 al 14%, oltre a superinfezioni da E. faecalis, germe non suscettibile al quinupristin/dalfopristin.
Farmacocinetica
Dal punto di vista farmacocinetico, dopo singola somministrazione endovenosa l'emivita plasmatica del quinupristin e del dalfopristin è di circa 1 ora e 0,6 ore rispettivamente; i due composti sono metabolizzati a livello epatico ed eliminati prevalentemente per via biliare (circa 75%). Sebbene l'AUC aumenti fino a 2-3 volte in pazienti con cirrosi e la clearance sia lievemente ridotta nei soggetti con grave insufficienza renale, il dosaggio del farmaco non necessita di aggiustamenti nei pazienti con insufficienza renale od epatica, in dialisi peritoneale, anziani, od obesi. I due composti sono metabolizzati dal sistema microsomiale P450, in particolare dal citocromo CYP3A4 la cui attività viene inibita competitivamente dal quinupristin/dalfopristin che, di conseguenza, riduce la clearance plasmatica di altri farmaci metabolizzati dallo stesso enzima, quali ciclosporina, nifedipina ed altri calcio-antagonisti, midazolam, statine, farmaci antiretrovirali, ed altri. L'uso concomitante di altri farmaci metabolizzati dal CYP3A4 ed in grado di determinare un allungamento dell'intervallo QT è rischioso.
Efficacia clinica e sicurezza
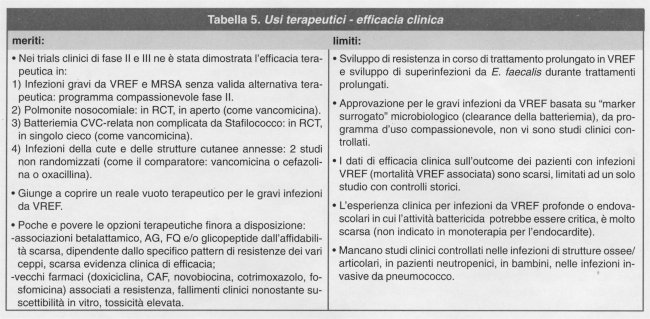
L'efficacia terapeutica del quinupristin/dalfopristin è stata dimostrata in studi clinici di fase II e III nei confronti di infezioni tipicamente sostenute da Gram-positivi; il farmaco si è dimostrato efficace soprattutto nel trattamento di patogeni Gram-positivi multiresistenti particolarmente problematici come S. aureus meticillino-resistente ed E. faecium vancomicino-resistente, oltre che nel trattamento empirico della polmonite nosocomiale e delle infezioni complicate della cute e dei tessuti molli4. In uno studio prospettico non comparativo di uso compassionevole in pazienti con infezioni gravi di vario genere sostenute da MRSA per le quali non esistevano alternative terapeutiche (fallimento o intolleranza ai glicopeptidi), quinupristin/dalfopristin ha mostrato una efficacia terapeutica clinico-batteriologica nel 71% dei pazienti5. Nel trattamento della batteriemia stafilococcica non complicata relata al catetere venoso centrale, quinupristin/dalfopristin è risultato altrettanto efficace della vancomicina in un piccolo studio, randomizzato, singolo-cieco6. L'efficacia del quinupristin/dalfopristin nel trattamento delle infezioni clinicamente significative da VREF è stata documentata in termini di successo microbiologico (eradicazione o clearance della batteriemia) nel 55% dei pazienti, generalmente con grave comorbidità, arruolati in uno studio prospettico non randomizzato nell'ambito di un protocollo di uso compassionevole7. L'efficacia comparativa del nuovo farmaco in termini di sopravvivenza globale, rispetto alle combinazioni di antibiotici finora utilizzabili per questo genere di infezioni problematiche è tuttavia supportata, ad oggi, solo da uno studio molto piccolo non randomizzato che ha utilizzato controlli storici8. Nel trattamento empirico di combinazione (con aztreonam) della polmonite nosocomiale, quinupristin/dalfopristin è risultato efficace quanto la vancomicina in uno studio clinico randomizzato, in aperto9. In due studi multicentrici10 nel trattamento delle infezioni della cute e dei tessuti molli non complicate, quinupristin/dalfopristin ha mostrato una efficacia clinica simile a quella dei farmaci comparatori tradizionalmente utilizzati per questa indicazione (vancomicina, oxacillina, cefazolina). Oltre alle infezioni sopramenzionate, in cui la validità del farmaco è stata formalmente valutata nell'ambito di trials clinici, descrizioni di singoli casi clinici o di piccole serie di pazienti hanno riportato l'efficacia del quinupristin/dalfopristin anche nel trattamento di artriti/borsiti settiche, osteomieliti, peritoniti relate a dialisi peritoneale, empiemi, infezioni endovascolari (protesi valvolari, impianti aortici) per lo più sostenute da S. aureus, stafilococchi coagulasi-negativi, enterococchi (anche vancomicino-resistenti) o streptococchi.
Nei vari studi clinici, gli effetti indesiderati associati all'uso del quinupristin/dalfopristin sono di entità lieve-moderata in circa l'80% dei casi, ma sono stati di entità tale da indurre la sospensione del trattamento nel 15-19% dei pazienti, frequenza generalmente superiore a quella dei farmaci di confronto. Un effetto indesiderato particolarmente frequente (35-74%) è l'irritazione venosa nel punto di infusione (dolore o flebite) quando il farmaco viene somministrato attraverso una via periferica; l'utilità di aumentare il volume di infusione per minimizzare tale evento non è comprovata e può rendersi spesso necessario il ricorso ad un catetere venoso centrale per la continuazione del trattamento. Un altro effetto indesiderato che può risultare fastidioso con l'uso del quinupristin/dalfopristin è l'insorgenza di una sindrome artralgico-mialgica, la cui frequenza è globalmente relativamente bassa (1,3%), ma che nella popolazione dei pazienti più defedati o con grave comorbidità può raggiungere il 2,5-30,7% dei casi. Tale sindrome è reversibile, non associata a danno muscolare, in genere lieve-moderata, sebbene in alcuni pazienti anche molto grave da causare interruzione del trattamento (fino al 4% dei casi). Altri effetti indesiderati riportati sono nausea (4,6%), vomito (2,7%), diarrea (2,7%), rash cutaneo (2,5%), incremento della bilirubina diretta (3,1%).
Il quinupristin/dalfopristin è stato utilizzato negli studi clinici al dosaggio di 7,5 mg/kg, e.v. (infusione di 1 ora) ogni 8 ore nel trattamento delle infezioni gravi da VREF e MRSA e nella polmonite nosocomiale; ogni 8 o 12 ore nel trattamento delle infezioni complicate della cute e dei tessuti molli.
Ruolo terapeutico
Con l'approvazione del quinupristin/dalfopristin si aggiunge al repertorio dei farmaci antibatterici una ulteriore importante risorsa. In un'era di crescente pressione ecologica di selezione per il massiccio uso di antibiotici di ogni classe e di inesorabile diffusione clinica di ceppi batterici dotati contemporaneamente di resistenza verso molteplici classi di agenti antibatterici, solo dall'arrivo di un composto appartenente ad una classe chimica del tutto nuova o quasi ci si può attendere un reale e duraturo vantaggio rispetto all'esistente in termini di spettro di attività sui patogeni in circolazione, e soprattutto su quelli oggi più problematici. Effettivamente il sostanziale ed innovativo merito del quinupristin/dalfopristin rispetto agli antibiotici già a disposizione è la sua quasi uniforme attività inibitoria contro i cocchi Gram-positivi multiresistenti emergenti ed in via di preoccupante diffusione (VREF, MRSA, PRSP). Per il trattamento delle gravi infezioni batteriemiche da VREF, patologia nosocomiale in aumento tra i pazienti più critici, questo farmaco giunge a coprire una reale necessità terapeutica per la quale può essere considerato di scelta, dato che le attuali opzioni di trattamento appaiono limitate, aleatorie, di efficacia non comprovata ed in alcuni casi gravate da tossicità elevata. Il nuovo farmaco si affianca ai glicopeptidi per il trattamento delle infezioni da MRSA, costituendo una preziosa alternativa nei pazienti intolleranti o scarsamente responsivi alla vancomicina / teicoplanina. Va sottolineato comunque che per questo genere di infezioni il ruolo del quinupristin/dalfopristin deve misurarsi con quello del linezolid, farmaco appartenente ad una classe del tutto nuova di antibatterici (oxazolidinoni), di più recente introduzione (vedi oltre). Sebbene non esistano ad oggi studi di confronto diretto tra i due farmaci, un profilo di tollerabilità ed interazioni potenzialmente più favorevole, nonché una superiore maneggevolezza nella possibilità di prosecuzione orale del trattamento potrebbero rendere preferibile il linezolid come prima linea per queste indicazioni. L'esperienza clinica con il quinupristin/dalfopristin è ancora molto scarsa per tutta una serie di infezioni gravi da cocchi Gram-positivi per le quali può essere teoricamente critica una elevata attività battericida (osteomieliti, infezioni profonde addominali, infezioni di materiale protesico, infezioni endovascolari, endocarditi, infezioni invasive da pneumococco), così come per specifiche popolazioni di pazienti (bambini, neutropenici, ecc.), mancando completamente studi di fase II o III pertinenti. In definitiva quindi, oltre al prezioso ruolo contro i patogeni multiresistenti sopramenzionati (VREF e MRSA), per tutte le altre infezioni anche gravi sostenute da ceppi comuni di Gram-positivi, per le quali esistono valide alternative terapeutiche, la possibile efficacia del quinupristin/dalfopristin deve essere pesata contro l'esperienza clinica (da RCT) ancora limitata con il farmaco, un profilo di tossicità ed interazioni farmacologiche probabilmente non favorevole, e, non ultimo, il rischio concreto che un uso allargato e sconsiderato di questo nuovo farmaco favorisca la precoce emergenza di ceppi resistenti, riducendone il periodo di utilità clinica o inficiando largamente per il futuro le potenzialità di ulteriori composti della nuova classe al momento in fase di sviluppo. L'impiego del quinupristin/dalfopristin per le comuni infezioni da Gram-positivi ed in "setting" empirico andrebbe perciò limitato rigorosamente. Le Tabelle 3, 4, 5, riassumono i punti più rilevanti.
Struttura e attività
Dal punto di vista strutturale il linezolid (Zyvoxid, Pharmacia) è il primo rappresentante di una classe del tutto nuova di agenti antibatterici di sintesi: gli oxazolidinoni. I composti di questa classe inibiscono la sintesi proteica batterica a livello ribosomiale per interferenza con la formazione del complesso di iniziazione t-RNA ribosoma, meccanismo d'azione unico e peculiare non suscettibile di resistenza crociata con gli altri agenti tradizionali inibitori della sintesi proteica. Lo spettro di attività antibatterica del linezolid è sostanzialmente limitato ai Gram-positivi, compresi i cocchi Gram-positivi emergenti resistenti a penicillina, cefalosporine, oxacillina e vancomicina. Il linezolid inibisce S. aureus sia meticillino-sensibile che resistente (MRSA) e dati preliminari indicano che il farmaco è attivo anche sui ceppi con sensibilità intermedia ai glicopeptidi (GISA). E' attivo sugli stafilococchi coagulasi-negativi anche meticillino-resistenti e sugli enterococchi, sia E. faecium che E. faecalis, compresi i ceppi vancomicino-resistenti (VREF). S. pyogenes, streptococchi viridanti e S. pneumoniae, compresi i ceppi resistenti a penicillina e cefalosporine (PRSP), sono suscettibili al linezolid. Il farmaco è inoltre attivo su Bacillus spp., Corynebacterium spp., Listeria monocytogenes, Rhodococcus equi. Alcuni anaerobi sono suscettibili come: Clostridium perfringens, C. difficile, Peptostreptococcus,Propionibacterium acnes, Bacteroides fragilis e Fusobacterium. Il linezolid è invece scarsamente attivo o inattivo sui bacilli Gram-negativi come Haemophilus influenzae, Moraxella catarrhalis, Legionella pneumophila, enterobacteriacee e P. aeruginosa. Sebbene sia stato riscontrato un certo grado di attività su Chlamydia e Mycoplasma, i dati di suscettibilità al linezolid di tali agenti respiratori atipici sono ancora limitati e sono già stati descritti ceppi di Mycoplasma pneumoniae del tutto resistenti. Il linezolid mostra invece una buona attività inibitoria su svariate specie di micobatteri, compresi M. tubercolosis, anche multiresistente, Mycobacterium avium-complex e micobatteri a rapida crescita. Il linezolid è generalmente batteriostatico; presenta un effetto post-antibiotico in vivo contro S. aureus e S. pneumoniae. Sebbene una resistenza al farmaco sia difficilmente inducibile in vitro, resistenza in corso di terapia prolungata è stata documentata in E. faecium ed E. faecalis, anche se come evento raro, in alcuni pazienti trattati nell'ambito del programma d'uso compassionevole e portatori di ascessi profondi non drenati o di materiali protesici infetti non rimossi. E' inoltre già stata descritta la resistenza al linezolid anche in un isolato clinico di S. aureus causa di peritonite in un paziente in dialisi peritoneale trattato con il farmaco.
Farmacocinetica
Il linezolid possiede un profilo farmacocinetico favorevole in quanto viene rapidamente (Cmax: 1-2 ore) e completamente assorbito per via orale con una biodisponibilità del 100%, non influenzata dai pasti. Può quindi essere somministrato indifferentemente per via endovenosa ed orale allo stesso dosaggio. L'emivita di eliminazione è di 4,5-5,5 ore. Il farmaco viene metabolizzato estesamente per ossidazione dell'anello morfolinico, quindi eliminato sia per via renale che biliare; il citocromo P450 non sembra essere coinvolto nella sua biotrasformazione e non è richiesto alcun aggiustamento di dosaggio nei pazienti con insufficienza renale lieve-moderata (clearance della creatinina: 10-80 ml/min) o insufficienza epatica moderata. Il farmaco viene dializzato per cui è richiesto un supplemento di dose dopo emodialisi.
Efficacia clinica e sicurezza
L'efficacia terapeutica del linezolid è stata valutata in alcuni studi clinici randomizzati di fase III nel trattamento delle infezioni sostenute da patogeni Gram-positivi antibiotico-resistenti (MRSA e VREF), della polmonite nosocomiale, della polmonite acquisita in comunità, delle infezioni della cute e dei tessuti molli complicate e non complicate11. Tuttavia buona parte di questi studi clinici sono stati pubblicati solamente come abstracts. In uno studio clinico randomizzato in aperto12 il linezolid è risultato altrettanto efficace della vancomicina nel trattamento delle infezioni gravi da MRSA (prevalentemente polmoniti, infezioni della cute e dei tessuti molli ed infezioni urinarie). Il farmaco, al dosaggio di 600 mg b.i.d., ha inoltre dimostrato efficacia clinica (67%) nel trattamento delle infezioni causate da VREF (prevalentemente batteriemie, infezioni cutanee e delle vie urinarie)13. Nel trattamento di combinazione (con aztreonam) della polmonite nosocomiale, il linezolid è risultato altrettanto efficace della vancomicina in uno studio clinico controllato, in doppio cieco14. Nel trattamento della polmonite acquisita in comunità il linezolid è risultato efficace quanto il cefpodoxime in pazienti ambulatoriali arruolati in uno studio randomizzato, in singolo cieco15, mentre in un altro studio in aperto in pazienti ospedalizzati è risultato altrettanto efficace del ceftriaxone seguito o meno dal cefpodoxime orale16. L'efficacia del linezolid è risultata pari a quella della claritromicina nelle infezioni della cute e dei tessuti molli non complicate, e pari a quella della oxacillina/dicloxacillina in quelle complicate, in due studi clinici randomizzati, doppio cieco17,18. Descrizioni pubblicate di singoli casi clinici o di piccole serie di pazienti hanno riportato successi terapeutici con il linezolid nel trattamento di endocarditi infettive, osteomieliti e meningiti sostenute da patogeni Gram-positivi multiresistenti, ma l'evidenza di efficacia in questo tipo di infezioni o nei pazienti immunocompromessi è ancora molto limitata.
I risultati degli studi di fase III suggeriscono un buon profilo di tollerabilità globale del linezolid, con la necessità di interruzione del trattamento per effetti indesiderati nel 3-4% dei casi. Gli effetti indesiderati più frequenti sono: diarrea (circa 8%), nausea (circa 6%) e cefalea (circa 6%); rash cutaneo si è manifestato nel 2,5% dei casi. L'effetto indesiderato più grave e peculiare verificatosi nei trials clinici è stata la trombocitopenia verificatasi mediamente nel 2,4% dei casi. L'incidenza della trombocitopenia è dipendente dalla durata del trattamento, divenendo più probabile per terapie oltre le 2 settimane. Si tratta comunque di un effetto di soppressione midollare reversibile alla sospensione del farmaco, così come la possibile leucopenia ed anemia, che tuttavia comporta una certa cautela ed un adeguato monitoraggio nella somministrazione degli oxazolidinoni a pazienti neutropenici, post-trapianto di midollo osseo, con diatesi emorragiche, con precedente trombocitopenia, o che assumono altri farmaci che possono interferire con la conta e la funzione piastrinica. Dal momento che il linezolid è un inibitore reversibile delle monoaminoossidasi (MAO), anche se debole, non essendo ancora completamente delineata la rilevanza clinica della sua attività anti-MAO, viene sconsigliata l'assunzione concomitante di grandi quantità di alimenti ad elevato contenuto di tiramina, sebbene negli studi clinici non siano mai stati riportati eventi avversi ascrivibili alla potenziale inibizione dell'enzima. Per lo stesso motivo, il linezolid ha, potenzialmente, interazioni farmacologiche con agenti adrenergici, dopaminergici e serotoninergici, dovute, ad esempio, ad una possibile accentuazione reversibile degli effetti pressori di adrenalina e dopamina, o ad una possibile facilitazione di una sindrome serotoninergica in caso di impiego concomitante di antidepressivi inibitori specifici del reuptake della serotonina (SSRI). Nonostante nei pazienti arruolati negli studi clinici con linezolid che assumevano anche tali antidepressivi non sia mai stata riportata una sindrome serotoninergica, l'esperienza clinica con tale associazione è ad oggi molto limitata, per cui essa andrebbe effettuata con molta cautela ed attenzione.
Il linezolid è stato utilizzato negli studi clinici al dosaggio di 600 mg ogni 12 ore, per 7-28 giorni nel trattamento delle infezioni da VREF e MRSA, per 10-14 giorni nella polmonite nosocomiale o acquisita in comunità e nelle infezioni complicate della cute e dei tessuti molli; al dosaggio di 400 mg ogni 12 ore, per 10-14 giorni, nelle infezioni non complicate della cute e dei tessuti molli. Il farmaco è disponibile sia in formulazione endovenosa che orale, senza necessità di variare il dosaggio passando dall'uno all'altro regime terapeutico.
Ruolo terapeutico
Con l'approvazione del linezolid si aggiunge all'armamentario antibiotico una ulteriore risorsa significativa ed innovativa in quanto, similmente al quinupristin/dalfopristin, il farmaco è il primo di una classe del tutto nuova di antibatterici di sintesi, con gli attesi vantaggi che ciò implica in termini di scarse resistenze attese tra i patogeni in circolazione selezionati da decenni di pesante pressione ecologica, e come tale può offrire un'attività inibitoria peculiare e rimarchevole sui cocchi Gram-positivi multiresistenti oggi emergenti (VREF, MRSA, GISA, PRSP). Per il trattamento delle infezioni gravi e batteriemiche da VREF, similmente a quanto detto per il quinupristin/dalfopristin ed affiancandosi ad esso, il linezolid viene a corroborare le poche opzioni terapeutiche finora disponibili, anzi, la sua attività comprendente anche E. faecalis e la sua maggiore maneggevolezza (profilo di tollerabilità buono e disponibilità anche di una formulazione orale) potrebbero rapidamente rendere il linezolid farmaco di prima scelta per questa indicazione, in assenza di studi di confronto diretto con quinupristin/dalfopristin. Il linezolid sembra poter avere un importante ruolo terapeutico anche nel trattamento delle infezioni da MRSA, come alternativa ai glicopeptidi, nei pazienti intolleranti alla vancomicina o in quelli scarsamente responsivi ai glicopeptidi per infezioni da ceppi con sensibilità ridotta (MIC elevate, resistenza eterogenea a vancomicina/teicoplanina). Un ruolo ulteriore nelle infezioni da MRSA, ed unico del linezolid, reso possibile dalla sua ottima biodisponibilità orale, riguarda sia la possibilità di un rapido "switch" orale di una terapia parenterale con glicopeptide in pazienti con pronta risposta clinica, in maniera da favorire la dimissione precoce e ridurre i rischi connessi con prolungate ospedalizzazioni o con il mantenimento a domicilio di cateteri venosi centrali a dimora, sia l'agevole cura di quei pazienti con infezioni da MRSA candidati ad un trattamento ambulatoriale a lungo termine (es. osteomieliti). A questo proposito va comunque sottolineato che i dati più attendibili di tollerabilità e sicurezza a lungo termine del linezolid sono ancora scarsi in quanto negli studi clinici la durata massima dei trattamenti è stata di quattro settimane e l'incidenza di quello che sembra essere il principale effetto indesiderato limitante il trattamento (depressione dell'emopoiesi) appare essere durata-dipendente.
Mancano inoltre studi clinici di fase II o III sulla efficacia del linezolid nelle infezioni delle strutture osteo-articolari che richiedono tipicamente trattamenti prolungati e che potrebbero avvalersi dello spettro di attività e della formulazione orale del nuovo farmaco. L'efficacia del linezolid, farmaco sostanzialmente batteriostatico, deve essere ancora validata da studi clinici in quelle infezioni come l'endocardite (da MRSA o enterococco) per le quali tradizionalmente una elevata attività battericida viene considerata di importanza critica. Il linezolid ha le caratteristiche per poter svolgere in futuro un ruolo probabilmente importante nelle infezioni da M. tuberculosis multiresistente. Andrebbe sottolineato che il farmaco ha ricevuto anche l'approvazione per il trattamento di comuni infezioni sostenute dagli usuali patogeni Gram-positivi comunitari, o presunte tali, come la polmonite acquisita in comunità e le infezioni non complicate della cute e dei tessuti molli. Tuttavia, anche in considerazione del fatto che la resistenza al linezolid sotto pressione di selezione in corso di trattamento è già stata documentata, l'uso empirico del nuovo farmaco per tali infezioni per le quali esistono numerose, valide (e meno costose) alternative terapeutiche (rispetto alle quali il linezolid non si è dimostrato superiore) appare inappropriato ed andrebbe scoraggiato. Sebbene recenti linee guida sul trattamento della polmonite acquisita in comunità inseriscano il linezolid come possibile opzione in pazienti con polmonite pneumococcica grave allergici ai beta-lattamici, l'esperienza clinica specifica (da RCT) con il linezolid per questa indicazione è ancora molto scarsa. Un massiccio ed "iper-entusiastico" uso del linezolid in setting extraospedaliero favorito dalla buona biodisponibilità orale e dalla buona tollerabilità condurrebbe inevitabilmente alla precoce emergenza di patogeni resistenti non solo al nuovo farmaco, ma anche ai possibili futuri oxazolidinoni con caratteristiche migliorate, attualmente in fase di sviluppo. L'impiego empirico del linezolid per le banali infezioni sostenute presuntivamente dai comuni cocchi gram-positivi andrebbe rigorosamente controllato e limitato. Le Tabelle 6, 7, 8, riassumono i punti più rilevanti.
Struttura e attività
La telitromicina (Ketek, Lepetit) è il primo agente antibatterico appartenente ad una nuova sottoclasse del gruppo degli antibiotici macrolidi-lincosamidi-streptogramine (MLS): i ketolidi. Ha in comune con l'eritromicina l'anello lattonico a 14 atomi, con peculiari sostituzioni, un chetone in posizione 3 (da cui la trasformazione della molecola da macrolide a ketolide) ed un carbammato ciclico in posizione 11-12, che conferiscono alla nuova molecola una maggiore attività antibatterica sui patogeni esprimenti i comuni meccanismi di resistenza ai macrolidi ed al gruppo MLS in generale (mutazioni del sito di legame ribosomiale con riduzione dell'affinità per l'antibiotico, pompa di estrusione attiva, inattivazione enzimatica). Questa capacità del ketolide di superamento della macrolido-resistenza non sembra essere tuttavia generalizzata, ma variabile secondo le specie batteriche, essendo pressoché uniformemente presente in S. pneumoniae, meno uniforme in S. pyogenes, ridotta in S. aureus e quasi nulla negli enterococchi eritromicino-resistenti. Il meccanismo d'azione antibatterica ed il sito di legame della telitromicina sono gli stessi dei macrolidi, ovvero la inibizione della sintesi proteica e dell'assemblaggio del ribosoma dopo legame a due distinti domini della subunità 50S, ma l'affinità di legame della telitromicina è 6 e 10 volte maggiore rispetto alla claritromicina ed all'eritromicina rispettivamente. In sostanza lo spettro di attività antibatterica è simile a quello dei macrolidi, comprendente in particolare tutti i principali patogeni respiratori normalmente contratti in comunità, ma con una conservata attività verso la maggior parte dei cocchi Gram-positivi macrolide-resistenti. Contro i Gram-negativi (H. influenzae e Moraxella), agenti atipici e micobatteri, l'attività della telitromicina è invece sovrapponibile a quella dei comuni macrolidi. In particolare, la telitromicina è attiva in vitro su S. aureus meticillino-sensibile e sugli stafilococchi coagulasi-negativi meticillino-sensibili, compresi alcuni dei ceppi eritromicino-resistenti (circa il 25% di essi), su S. pneumoniae, uniformemente anche sui ceppi eritromicino- e penicillino-resistenti, su S. pyogenes anche eritromicino-resistente (pur se meno uniformemente che per lo pneumococco), sugli streptococchi viridanti, sugli enterococchi eritromicino-sensibili ma non sui ceppi eritromicino-resistenti (come per i macrolidi, ne è comunque sconsigliabile l'uso nelle infezioni clinicamente significative da enterococco), su Listeria monocytogenes. La telitromicina inibisce inoltre H. influenzae (lievemente più attivo della claritromicina e meno attivo della azitromicina) e Moraxella catarrhalis, anche beta-lattamasi produttori, N. meningitidis, N. gonorrhoeae, Legionella pneumophila, alcuni anaerobi come Peptostreptococcus spp,Actinomyces spp e C. perfringens, gli agenti "atipici" Chlamydia pneumoniae e trachomatis, Mycoplasma pneumoniae, Ureaplasma urealyticum, anche doxiciclino-resistente. La telitromicina non è invece attiva su enterobacteriacee, P. aeruginosa, Acinetobacter, Bacteroides, Fusobacterium, Corynebacterium JK, Mycoplasma hominis, M. tuberculosis. Similmente ai macrolidi, la telitromicina esercita un prolungato effetto post-antibiotico nei confronti dei comuni patogeni respiratori ed è battericida nei loro confronti a concentrazioni da 2 ad 8 volte le MIC.
Farmacocinetica
La telitromicina dopo somministrazione orale ha un assorbimento rapido con picco di concentrazione plasmatica in 1-2 ore, non influenzato dai pasti. La biodisponibilità orale del farmaco è del 57%, essendo la molecola sottoposta ad un esteso metabolismo epatico di primo passaggio (per il 33%); la penetrazione e le concentrazioni tessutali sono elevate soprattutto nei tessuti e fluidi delle vie respiratorie superiori ed inferiori e nei fagociti. Il farmaco viene per gran parte metabolizzato a livello epatico, in uguale misura attraverso il citocromo P450 (CYP3A4 e CYP2D6) ed altre vie enzimatiche indipendenti da esso, quindi eliminato prevalentemente per via biliare (75%) ed in minor misura per via urinaria (17%). L'emivita plasmatica è di circa 9-10 ore e non sembra necessario nessun aggiustamento di dose in pazienti con insufficienza epatica o renale e negli anziani. Dal momento che il farmaco è un substrato ed un inibitore del citocromo P450, la sua concentrazione plasmatica può essere aumentata da altri composti inibitori dell'enzima, e la telitromicina può aumentare a sua volta quella di altri farmaci metabolizzati dal CYP3A4 e CYP2D6 (statine, benzodiazepine, aminofillina e digossina); tuttavia gli studi riguardanti le interazioni metaboliche della telitromicina con altri specifici farmaci sono ad oggi ancora limitati, per cui la somministrazione contemporanea di qualsiasi farmaco metabolizzato in particolare dal CYP3A4 va considerata come potenzialmente pericolosa ed andrebbe effettuata con le dovute cautele. Come gli altri macrolidi, anche il ketolide va considerato potenzialmente capace di indurre prolungamento dell'intervallo QT e la co-somministrazione di altri farmaci con la stessa caratteristica, particolarmente se substrati del CYP3A4, è rischiosa.
Efficacia clinica e sicurezza
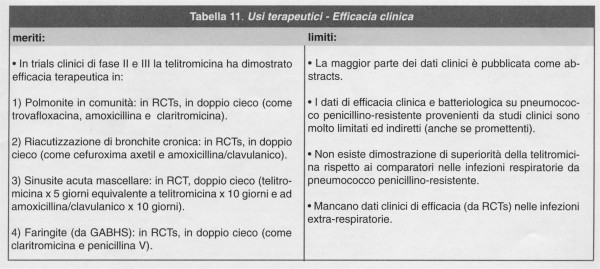
Finora, l'efficacia terapeutica della telitromicina è stata valutata in studi clinici randomizzati di fase III soltanto nel trattamento delle infezioni delle alte e basse vie aeree: polmonite acquisita in comunità, bronchite cronica riacutizzata, sinusite acuta mascellare, faringo-tonsillite19. Tuttavia quasi tutti questi studi clinici comparativi, che avevano generalmente come obiettivo la dimostrazione di "non inferiorità" della telitromicina rispetto al trattamento consolidato, sono stati pubblicati ad oggi solamente come abstracts e presentati a congressi (tranne uno di confronto con penicillina V nella faringo-tonsillite). Nel trattamento della polmonite acquisita in comunità la telitromicina è risultata efficace sul piano clinico e microbiologico quanto l'amoxicillina e la claritromicina, in due studi clinici randomizzati, in doppio cieco, in cui la gran parte dei pazienti arruolati presentava una polmonite di gravità lieve-moderata, tipicamente non richiedente l'ospedalizzazione20,21. Nei pazienti con polmonite grave, i dati di efficacia, seppure promettenti, sono alquanto limitati. Inoltre, nonostante le caratteristiche di attività antibatterica del nuovo farmaco sembrino supportare una sua superiorità terapeutica nei confronti dei patogeni respiratori antibiotico-resistenti, con particolare riguardo a S. pneumoniae, in realtà finora i dati clinici disponibili relativi al trattamento con telitromicina di polmoniti da pneumococco penicillino-resistente, macrolide-resistente, o resistente ad entrambe, sono estremamente scarsi (3 isolati, 1 isolato ed 1 isolato rispettivamente, tutti batteriemici). Nella bronchite cronica riacutizzata, un trattamento abbreviato (5 giorni) con telitromicina è risultato altrettanto efficace del trattamento orale standard di 10 giorni con amoxicillina/clavulanato e con cefuroxime in due studi clinici randomizzati, in doppio cieco22,23. Anche per la sinusite acuta mascellare, lo stesso ciclo di trattamento breve di 5 giorni con telitromicina si è dimostrato altrettanto efficace di un trattamento di durata convenzionale di 10 giorni con lo stesso farmaco o con amoxicillina/clavulanato orale, in un altro studio clinico randomizzato, in doppio cieco24. La telitromicina si aggiunge quindi agli altri antibiotici (es. azitromicina, cotrimoxazolo, cefpodoxime e cefixime) con i quali un trattamento abbreviato (5 giorni, o meno) della sinusite acuta si è dimostrato adeguato quanto un trattamento convenzionale di 10-14 giorni. Nel trattamento della faringo-tonsillite da streptococco b-emolitico di gruppo A, un ciclo di trattamento abbreviato (5 giorni) con telitromicina ha dimostrato simile efficacia clinica e microbiologica di un trattamento standard di 10 giorni con penicillina V o con claritromicina nel corso di due studi clinici randomizzati, in doppio cieco25,26. Sebbene la penicillina V non sia consigliabile per trattamenti abbreviati della faringo-tonsillite da streptococco beta-emolitico di gruppo A, altri farmaci già a disposizione come cefpodoxime ed azitromicina, similmente alla telitromicina hanno dimostrato efficacia per questo genere di indicazione.
Il profilo di tollerabilità della telitromicina emerso dagli studi clinici è sostanzialmente buono: i pazienti che hanno dovuto interrompere il trattamento per effetti indesiderati sono pochi. I più comuni sono stati quelli gastroenterici; in particolare diarrea (14,4%) e nausea (9%), generalmente più frequenti con telitromicina che con i comparatori (macrolidi o betalattamici, ma meno frequenti rispetto ad amoxicillina/clavulanico). Altri effetti indesiderati riportati sono stati cefalea e vertigini (fino al 6%), offuscamento visivo (raro e transitorio, ma di incerto significato clinico), alterazioni lievi degli indici di funzionalità epatica. La sicurezza della telitromicina in gravidanza non è nota. Recentemente sono stati segnalati casi di riacutizzazione di miastenia grave, incluso un caso di decesso, in pazienti con miastenia grave accertata che avevano assunto telitromicina per il trattamento delle infezioni del tratto respiratorio. La riacutizzazione della debolezza muscolare, dispnea o grave insufficienza respiratoria acuta si sono manifestate entro poche ore dalla prima somministrazione del farmaco. Il meccanismo di tali riacutizzazioni non è noto. L'insufficienza respiratoria acuta in pazienti con miastenia grave può metterne in pericolo la vita. In caso di riacutizzazione di tali sintomi, il farmaco deve essere interrotto e deve essere istituito un adeguato trattamento di supporto come indicato dalla situazione clinica. La telitromicina non dovrebbe essere somministrata a pazienti affetti da miastenia grave a meno che non siano disponibili terapie alternative. Quando si inizia un trattamento con telitromicina, i pazienti affetti da miastenia grave dovranno essere attentamente controllati; pazienti con miastenia grave dovranno essere informati di contattare immediatamente un medico se si accorgono di qualsiasi peggioramento della loro sintomatologia dopo aver assunto il farmaco.
Il profilo farmacocinetico e la buona penetrazione tessutale della telitromicina permettono la monosomministrazione giornaliera e cicli di trattamento abbreviati rispetto alla durata standard; il dosaggio utilizzato negli studi clinici è 800 mg una volta al giorno, per via orale. Non è disponibile ad oggi una formulazione parenterale, che è in corso di sviluppo.
Ruolo terapeutico
L'efficacia clinica della telitromicina è stata valutata in studi clinici di fase III esclusivamente nelle infezioni respiratorie ed il nuovo farmaco è in corso di registrazione per il trattamento orale di queste affezioni. In questa area della antibioticoterapia, già affollata di concorrenti (penicilline protette, cefalosporine, nuovi fluorochinoloni, macrolidi/azalidi, ecc.) il vantaggio principale che questo ketolide sembra apportare è rappresentato sostanzialmente dalla sua migliorata attività inibitoria sullo pneumococco macrolide-resistente e/o penicillino-resistente. L'attività del ketolide sugli altri patogeni respiratori tipici ed atipici è sovrapponibile a quella della classe dei macrolidi/azalidi già in uso da tempo, e l'affidabilità dell'attività su H. influenzae merita probabilmente ulteriori prove di efficacia da studi clinici randomizzati. Sebbene le caratteristiche farmacodinamiche della telitromicina configurino il nuovo farmaco come un'arma utile specificamente per le infezioni respiratorie da pneumococco antibiotico-resistente, i dati di efficacia clinico-batteriologica nelle gravi infezioni da PRSP derivanti dagli studi clinici sono estremamente scarsi ed indiretti e nessuno studio clinico con la telitromicina è stato effettuato sulle infezioni da PRSP. D'altra parte nel trattamento empirico delle infezioni respiratorie la telitromicina si è dimostrata di uguale efficacia e non superiore ai macrolidi (claritromicina) o ai beta-lattamici già in uso da tempo, mentre, semmai il suo profilo di tollerabilità è apparso essere meno favorevole rispetto al macrolide comparatore per quanto riguarda gli effetti gastrointestinali. Un vantaggio della telitromicina nella terapia di alcune infezioni respiratorie minori (sinusite acuta e faringotonsillite, ma non la polmonite) è rappresentato dalla dimostrata efficacia di un ciclo di trattamento abbreviato con il ketolide rispetto al trattamento di durata standard con i farmaci di confronto, tuttavia questa caratteristica è propria anche di alcuni beta-lattamici già in uso e, tra i macrolidi/azalidi, della stessa azitromicina che, rispetto alla telitromicina sembra avere tra l'altro un profilo di tollerabilità gastrointestinale e di potenziali interazioni più favorevole. Per il momento quindi il nuovo ketolide andrebbe probabilmente riservato al trattamento di quelle specifiche condizioni in cui viene isolato uno pneumococco macrolide-resistente o penicillino-resistente, o in cui questa evenienza è altamente sospetta sulla base di considerazioni clinico-epidemiologiche. Nel nostro paese la prevalenza dello pneumococco con piena resistenza alla penicillina rimane tuttora molto bassa (3-4%), sebbene la resistenza all'eritromicina sia decisamente più elevata (circa il 25% nel 1999) e la resistenza ai macrolidi tra i ceppi penicillino-resistenti può aggirarsi attorno al 50%. In futuro, se la penicillino- e macrolide-resistenza tenderà ulteriormente a diffondersi tra i ceppi di pneumococco, aumenterà verosimilmente anche il ruolo terapeutico della telitromicina nelle infezioni respiratorie, ma allo stato attuale un uso "entusiastico" e sconsiderato non porterebbe vantaggi terapeutici sostanziali, ponendo invece il rischio di precoce emergenza di resistenze verso l'intera nascente famiglia dei ketolidi. Le Tabelle 9, 10, 11, riassumono i punti più rilevanti.
Bibliografia 1. Gold HS, Moellering RC Jr.: Antimicrobial-drug resistance. N Engl J Med 1996; 335: 1445-53. 2. Amyes SGB: The rise in bacterial resistance. BMJ 2000; 320: 199-200. 3. Cassell GH, Mekalanos J. Development of antimicrobial agents in the era of new and reemerging infectious diseases and increasing antibiotic resistance. JAMA 2001; 285: 601-605. 4. Lamb HM, Figgitt DP, Faulds D. Quinupristin/dalfopristin. A review of its use in the management of serious Gram-positive infections. Drugs 1999; 58: 1061-97. 5. Drew RH, Perfect JR, Srinath L et al. Treatment of methicillin-resistant Staphylococcus aureus infections with quinupristin-dalfopristin in patients intolerant of or failing prior therapy. J Antimicrob Chemother 2000;46: 775-84. 6. Raad I, Bompart F, Hachem R. Prospective, randomized dose-ranging open phase II pilot study of quinupristin-dalfopristin versus vancomycin in the treatment of catheter-related staphylococcal bacteremia. Eur J Clin Microbiol Infect Dis 1999; 18: 199-202. 7. Moellering RC, Linden PK, Reinhardt J et al. The efficacy and safety of quinupristin/dalfopristin for the treatment of infections caused by vancomycin-resistant Enterococcus faecium. J Antimicrob Chemother 1999;44: 251-61. 8. Linden PK, Pasculle AW, McDevitt D et al. Effect of quinupristin/dalfopristin on the outcome of vancomycin-resistant Enterococcus faecium bacteraemia: comparison with a control cohort. J Antimicrob Chemother 1997;39 Suppl A: 145-51. 9. Fagon J, Patrick H, Haas DW et al. Treatment of gram-positive nosocomial pneumonia. Prospective randomized comparison of quinupristin/dalfopristin versus vancomycin. Am J Respir Crit Care Med 2000; 161(3 Pt 1): 753-62. 10. Nichols RL, Graham DR, Barriere SL et al. Treatment of hospitalized patients with complicated gram-positive skin and skin structure infections: two randomized, multicentre studies of quinupristin/dalfopristin versus cefazolin, oxacillin or vancomycin. J Antimicrob Chemother 1999; 44: 263-73. 11. Perry CM, Jarvis B: Linezolid. A review of its use in the management of serious Gram-positive infections.Drugs 2001; 61: 525-551. 12. Stevens DL, Herr D, Lampiris H et al. Linezolid versus vancomycin for the treatment of methicillin-resistant Staphylococcus aureus infections. Clin Infect Dis 2002; 34: 1481-90. 13. Pharmacia and Upjohn: Linezolid for the treatment of vancomycin-resistant enterococcal infections: a double-blind trial comparing 600 mg linezolid every 12 hours with 200 mg linezolid every 12 hours (study report M/1260/0054A). Peapack, NJ: Pharmacia and Upjohn 1999. 14. Rubinstein E, Cammarata S, Oliphant T et al. Linezolid (PNU-100766) versus vancomycin in the treatment of hospitalized patients with nosocomial pneumonia: a randomized, double-blind, multicenter study. Clin Infect Dis 2001; 32: 402-12. 15. Cammarata SK, Schueman LK, Timm JA et al. Oral linezolid in the treatment of community-acquired pneumonia : a phase III trial (abstract). Am J Resp Crit Care Med 2000; 161 (Ppt 2) Suppl: A654. 16. Cammarata SK, Bermudez M, Golin V et al. Comparison of linezolid versus ceftriaxone/cefpodoxime in the treatment of hospitalized community acquired pneumonia (abstract). 9th International Congress on Infectious Diseases 2000; Apr 10-13: Buenos Aires, 182. 17. Duvall SE, Sseas C, Bruss JB et al. Comparison of oral linezolid to oral clarithromycin in the treatment of uncomplicated skin infections: results from a multinational phase III trial (abstract). 9th International Congress on Infectious Diseases 2000; Apr 10-13: Buenos Aires, 181. 18. Stevens DL, Smith LG, Bruss JB et al. Randomized comparison of linezolid (PNU-100766) versus oxacillin-dicloxacillin for treatment of complicated skin and soft tissue infections. Antimicrob Agents Chemother 2000;44: 3408-13. 19. Bearden DT, Neuhauser MM, Garey KW: Telithromycin: an oral ketolide for respiratory infections.Pharmacotherapy 2001; 21: 1204-1222. 20. Hagberg L, Torres A, Van Rensburg D et al. Efficacy and tolerability of telithromycin (HMR 3647) vs. high-dose amoxicillin in the treatment of community-acquired pneumonia (abstr and poster). In: Program and abstracts of the 40th International Conference of Antimicrobial Agents and Chemotherapy. Washington, DC: American Society for Microbiology 2000: 490. 21. Tellier G, Hassman J, Leroy B et al. Oral telithromycin (HMR 3647; 800 mg od) is well tolerated and as effective as oral clarithromycin (500 mg bid) in community-acquired pneumonia (CAP) in adults (abstr and poster). In: Program and abstracts of the 40th International Conference of Antimicrobial Agents and Chemotherapy. Washington, DC: American Society for Microbiology 2000: 471. 22. Aubier M, Aldons P, Leak A et al. Efficacy and tolerability of a 5-day course of a new ketolide antimicrobial, telithromycin (HMR 3647), for the treatment of acute exacerbations of chronic bronchitis (AECB) in patients with COPD (abstr and poster). In: Program and abstracts of the 40th International Conference of Antimicrobial Agents and Chemotherapy. Washington, DC: American Society for Microbiology 2000: 489. 23. De Abate C, Heyder A, Leroy B et al. Oral telithromycin (HMR 3647; 800 mg od) for 5 days is well tolerated and as effective as cefuroxime axetil (500 mg bid) for 10 days in adults with acute exacerbations of chronic bronchitis (AECB) (abstr and poster). In: Program and abstracts of the 40th International Conference of Antimicrobial Agents and Chemotherapy. Washington, DC: American Society for Microbiology 2000: 471. 24. Tellier G, Lasko B, Leroy B et al. Oral telithromycin (HMR 3647; 800 mg od) for 5 days and 10 days is well tolerated and as effective as amoxicillin/clavulanic acid (500/125 mg tid) for 10 days in acute maxillary sinusitis (AMS) in adults (abstr and poster). In: Program and abstracts of the 40th International Conference of Antimicrobial Agents and Chemotherapy. Washington, DC: American Society for Microbiology 2000: 471. 25. Norrby SR, Rabie WJ, Bacart P et al. Efficacy of short-course therapy with the ketolide telithromycin compared with 10 days of penicillin V for the treatment of pharyngitis/tonsillitis. Scand J Infect Dis 2001; 33: 883-90. 26. Ziter P, Quinn J, Leroy B et al. Oral telithromycin (HMR 3647; 800 mg od) for 5 days is well tolerated and as effective as clarithromycin (250 mg bid) for 10 days in group A beta-hemolytic streptococcal pharyngitis/tonsillitis (abstr and poster). In: Program and abstracts of the 40th International Conference of Antimicrobial Agents and Chemotherapy. Washington, DC: American Society for Microbiology 2000: 472.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}