
L'introduzione dei nuovi antivirali per l'epatite C: è possibile un compromesso tra salute e mercato?


Quando si parla di innovazione in campo farmacologico, un esempio attuale che merita senz’altro di essere citato è quello dei nuovi farmaci per l’epatite C (HCV), che stanno cambiando le prospettive di cura per milioni di pazienti con epatite cronica . Parliamo in particolare della classe degli antivirali diretti (direct-acting antiviral, DAA) come sofosvubir, simeprevir e daclatasvir - tutti approvati per la commercializzazione in Europa nel 2014 - e delle associazioni precostituite di sofosbuvir + ledipasvir (appena approvato) o di ombitasvir/paritaprenavir/ritonavir associato a dasabuvir, di prossima commercializzazione in Europa. Altri farmaci sono attualmente oggetto di studi registrativi e anche di questi si può prevedere una prossima introduzione sul mercato. La novità di queste terapie è rappresentata dal fatto che, se pure con percentuali diverse di successo in base al genotipo virale e all’entità del danno epatico, il loro utilizzo – in base a vari schemi terapeutici (partendo dal singolo antivirale associato a ribavirina ed interferone possono arrivare a prevedere l’assenza di interferone e nel caso dell’associazione di due antivirali anche di ribavirina) - sembra determinare in una elevata percentuale di casi l’eradicazione del virus dopo 3-6 mesi di trattamento. Appare così ampiamente superato l’uso degli antivirali ad oggi disponibili per tale patologia (boceprevir e telaprevir, approvati da FDA ed EMA nel 2011 in associazione obbligata con interferone e ribavirina)1. Oltre ai dati che ne suggeriscono una maggiore efficacia rispetto agli standard terapeutici attuali, i nuovi antivirali possono avere importanti vantaggi in termini di tollerabilità, in particolare quando i regimi terapeutici consentono trattamenti più brevi senza associare né interferone né ribavirina2.
Quale efficacia nei diversi sottogruppi?
I risultati molto incoraggianti degli studi in termini di efficacia e sicurezza non dovrebbero tuttavia farci sorvolare su alcuni loro limiti che dovrebbero essere colmati da futuri studi. In particolare, esistono diversi genotipi virali su cui i farmaci hanno dimostrato un differente grado di efficacia, e un’ampia gamma di situazioni e di sottogruppi di pazienti (espressione di una diversa gravità clinica) che negli studi sono poco rappresentati. È il caso ad esempio dei pazienti più gravi con cirrosi (compensata o scompensata) o con fibrosi di grado elevato spesso valutati in sottogruppi di piccole dimensioni (di poche decine o anche meno pazienti). In alcuni casi inoltre le prove di efficacia ad oggi disponibili derivano esclusivamente da studi di fase II, che per definizione non hanno come obiettivo primario l’efficacia e che spesso non sono stati disegnati prevedendo una stratificazione rispetto al grado di fibrosi o di genotipi virali. Dettagli sui dati di efficacia, provenienti dagli studi randomizzati (RCT) disponibili, delle terapie con i nuovi antivirali con e senza interferone sono forniti da una revisione pubblicata dal BMJ nell’agosto del 20143.
Di seguito sono indicati i titoli delle tabelle della revisione dove sono presentati tali dati e i relativi link (ad accesso libero).
Risultati dei maggiori studi sulle terapie combinate antivirali diretti + peginterferon e ribarivina
Risultati dei maggiori studi sulle terapie con antivirali diretti senza interferoni
Diffusione dell’epatite C e costi delle terapie: un mix difficilmente sostenibile
La diffusione dell’epatite C è estremamente variabile nel mondo e può esserlo anche all’interno di ogni singolo Paese. Non potrebbe essere altrimenti considerando l’esistenza, sia attualmente sia nel passato, di standard precauzionali molto diversi nelle varie parti del mondo, ma anche nello stesso Paese fra Regione e Regione o addirittura fra comunità e comunità. Per tali ragioni in generale e nell’ambito di ogni singolo Paese non è facile avere stime affidabili dal punto di vista epidemiologico nelle varie sottopopolazioni, anche per il numero relativamente limitato di casistiche studiate. È peraltro noto come la prevalenza dell’infezione da HCV sia elevata in alcuni Paesi del Nord-Africa (come l’Egitto, dove è stimata addirittura nell’ordine del 15% della popolazione)4 e dell’Asia.
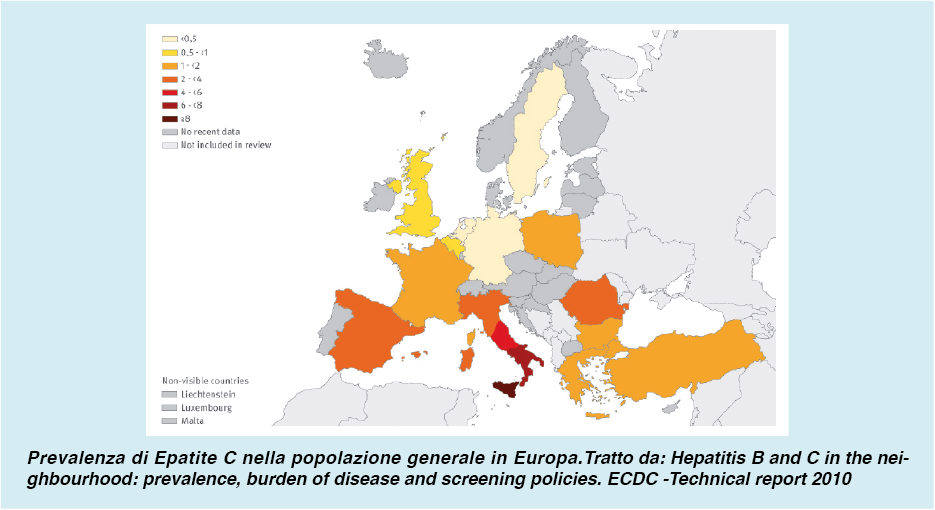 In Italia la variabilità delle stime disponibili consente solo di ipotizzare una prevalenza media attorno al 2% e un gradiente nord-sud (in alcune regioni meridionali la prevalenza sembra essere abbastanza superiore al dato medio nazionale)5. Anche la frequenza nella distribuzione dei vari genotipi non è omogenea; per l’Italia i pochi studi epidemiologici disponibili indicano range molto ampi, anche se tutti concordano che il genotipo 1 è quello prevalente6. Non è altresì chiara né la prevalenza né la distribuzione dei vari genotipi nelle varie popolazioni immigrate. Per quanto i dati di prevalenza siano incerti, anche considerando che un’ampia maggioranza di soggetti positivi al virus non è consapevole di esserlo (l’infezione ha un’evoluzione clinica lentissima e possono passare decenni prima che si manifestino dei sintomi), è molto probabile che il numero di pazienti potenzialmente eleggibili per una terapia in Italia sia nell’ordine delle centinaia di migliaia, e che il costo della terapia rappresenti un potenziale fattore limitante per le possibilità di trattamento. Senza considerare naturalmente che la possibilità – in linea del tutto teorica –di realizzare screening di massa, da alcuni proposta per individuare tutti i soggetti infetti, potrebbe portare alla luce numeri nettamente superiori.
In Italia la variabilità delle stime disponibili consente solo di ipotizzare una prevalenza media attorno al 2% e un gradiente nord-sud (in alcune regioni meridionali la prevalenza sembra essere abbastanza superiore al dato medio nazionale)5. Anche la frequenza nella distribuzione dei vari genotipi non è omogenea; per l’Italia i pochi studi epidemiologici disponibili indicano range molto ampi, anche se tutti concordano che il genotipo 1 è quello prevalente6. Non è altresì chiara né la prevalenza né la distribuzione dei vari genotipi nelle varie popolazioni immigrate. Per quanto i dati di prevalenza siano incerti, anche considerando che un’ampia maggioranza di soggetti positivi al virus non è consapevole di esserlo (l’infezione ha un’evoluzione clinica lentissima e possono passare decenni prima che si manifestino dei sintomi), è molto probabile che il numero di pazienti potenzialmente eleggibili per una terapia in Italia sia nell’ordine delle centinaia di migliaia, e che il costo della terapia rappresenti un potenziale fattore limitante per le possibilità di trattamento. Senza considerare naturalmente che la possibilità – in linea del tutto teorica –di realizzare screening di massa, da alcuni proposta per individuare tutti i soggetti infetti, potrebbe portare alla luce numeri nettamente superiori.
L’introduzione dei nuovi anti-HCV in Europa e in Italia
La commercializzazione di sofosvubir (il primo di questa classe ad arrivare in commercio) è stata approvata negli Stati Uniti il 6 dicembre 2013 e in Europa il 16 gennaio 2014. Già dall’ottobre 2013 EMA, su richiesta della Finlandia, aveva indicato i criteri per l’uso “compassionevole” del farmaco e cioè in pazienti:
1) in lista di attesa per trapianto epatico per prevenire una possibile reinfezione al momento del trapianto;
2) che dopo un trapianto epatico, avessero presentato una re-infezione aggressiva e con un alto rischio di morte o scompenso epatico nei successivi 12 mesi.
In Europa, sofosbuvir è attualmente disponibile in diversi Paesi fra cui Francia, Germania, Inghilterra, Spagna e Italia. Le rispettive Agenzie nazionali che hanno valutato/definito il valore aggiunto del farmaco hanno espresso pareri abbastanza disomogenei (vedi tabella) che riflettono le difficoltà sia nel definire la fascia di soggetti HCV positivi ai quali garantire prioritariamente il farmaco - considerando i costi esorbitanti del trattamento - sia le incertezze sui dati di efficacia ad oggi disponibili rispetto alle tipologie di pazienti (solo alcuni genotipi, alcuni livelli di fibrosi o di gravità clinica erano rappresentati in quantità numericamente accettabile negli studi) e alle terapie di confronto utilizzate negli RCT. In generale, viene sottolineata la necessità di definire dei criteri di priorità nell’accesso ai farmaci che tengano conto delle prove di efficacia, genotipo virale, gravità della malattia e risposta a precedenti cure.
In Italia è stata appena approvata la immissione in commercio di sofosbuvir dopo una negoziazione tra la ditta produttrice Gilead Sciences e AIFA durata quasi un anno durante la quale la stessa ditta, in modo irrituale rispetto alle regole vigenti, aveva accettato la richiesta di AIFA di rendere disponibile il farmaco per uso compassionevole per 1200 pazienti secondo i criteri suggeriti da EMA. La determina AIFA pubblicata il 5 dicembre scorso, che ufficializza la commercializzazione del farmaco in classe di rimborsabilità A PHT, gli attribuisce il requisito dell’innovatività terapeutica e i conseguenti benefici; la prescrizione a carico del SSN sarà limitata ai centri prescrittori individuati dalle regioni e avverrà tramite la compilazione di un piano terapeutico web-based che definisce i criteri di eleggibilità. Per quanto riguarda il prezzo invece la determina AIFA riporta esclusivamente quello ufficiale del farmaco (per 12 settimane di trattamento: prezzo ex factory:45.000 euro, prezzo al pubblico: 74.260 euro) e fa riferimento ad uno sconto obbligatorio alla strutture pubbliche sul prezzo ex factory e ad una durata del contratto di 18 mesi senza presentarne i particolari 7. Dal canto suo la ditta Gilead rispetto ai termini dell’accordo negoziale ha solo comunicato, in una lettera indirizzata alle direzioni di tutte le farmacie ospedaliere, che il costo per le strutture pubbliche della terapia completa con Sovaldi® sarà di 37.000 € (IVA esclusa) sia per la terapia con durata di 12, che di 24 o 48 settimane (o fino al trapianto). La Gilead ha inoltre comunicato la chiusura del programma di uso compassionevole dal 15 dicembre 2014. Secondo una strategia ben consolidata, la ditta preferisce non esplicitare i termini degli accordi per evitare che altri Paesi possano chiedere un prezzo analogo (nel caso questo sia inferiore a quello pagato altrove). La scheda di raccolta dati informatizzata AIFA indica i criteri di accesso al farmaco, facendo riferimento ad un ordine progressivo di priorità definito dalla CTS di AIFA in base all’urgenza clinica . In realtà non viene messo a disposizione alcun strumento per verificare il rispetto dell’ordine progressivo, quindi nella pratica tutti i pazienti che presentano uno dei 6 criteri definiti da AIFA potranno essere trattati.
I nuovi DAA: criticità dal punto di vista clinico, di accesso e gestionale
Il percorso che ha portato alla messa in commercio del sofosbuvir nel nostro paese ci porta ad esprimere una serie di considerazioni ed alcuni quesiti:
• dal punto di vista clinico: ai tempi molto lunghi nella messa a disposizione nel nostro paese del sofosbuvir (data la lunga durata della negoziazione sul prezzo) si aggiunge l’attuale indisponibilità di simeprevir (approvato da AIFA nel novembre 2014), di daclatasvir e dell’associazione sofosbuvir + ledipasvir, gli ultimi due già autorizzati per il commercio in Europa. Per un’altra associazione, ombitasvir/paritaprenavir/ritonavir + dasabuvir, di cui esiste già il parere positivo di EMA, sono stati definiti dall’AIFA alla fine di ottobre i criteri di accesso per un programma di uso compassionevole in accordo con la ditta produttrice AbbVie 8. In diversi casi questi farmaci permetteranno di evitare l’uso di interferone o anche di ribavirina, e potranno rappresentare un ulteriore avanzamento nella terapia dell’epatite C in termini di efficacia e di tollerabilità, evitando di investire risorse senza un ritorno clinico adeguato. Per alcuni pazienti, in particolare per quelli con il genotipo più frequente nel nostro paese (genotipo 1), sarà importante o addirittura fondamentale poter ricorrere alla associazione fra due diversi DAA per ottimizzare al massimo i trattamenti in termini di costo-efficacia. Tutti motivi, questi, per auspicare una rapida introduzione dei nuovi farmaci.
• dal punto di vista della possibilità di accesso a queste terapie: dati i prezzi indicati, per trattare alcune centinaia di migliaia di pazienti potenzialmente eleggibili occorrerebbero molti miliardi di euro, anche se l’entità esatta della spesa non è ad oggi ben quantificabile a causa dell’incertezza delle stime di prevalenza e soprattutto della mancata conoscenza delle “condizioni negoziali” attuali che definiscono gli sconti per l’acquisto del sofosbuvir da parte delle strutture pubbliche. Nulla ovviamente si sa di quali saranno i prezzi futuri dei numerosi DAA in arrivo anche se è prevedibile un loro progressivo calo. Per fronteggiare la attuale situazione è stato approvato un emendamento del Governo alla Legge di Stabilità 2015 che definisce l’istituzione di un fondo speciale per l’acquisto di farmaci innovativi, per l’ammontare complessivo di 1 miliardo di euro per i prossimi 2 anni9, che consentirà il trattamento con sofosbuvir e/o con gli altri DAA che stanno per essere commercializzati in base ai criteri identificati dall’AIFA per ognuno di essi. Il problema è che tale fondo sarà costituito per il 90% da risorse del Fondo Sanitario Nazionale (FSN), sottratte dunque ad altri usi. Inoltre, le regioni dovranno avere disponibilità economica per anticipare le spese per i trattamenti, che saranno rimborsati attraverso il fondo solo in un secondo momento. Si stanno delineando quindi due livelli diversi di problemi legati da un lato all’accesso immediato al farmaco/ai farmaci per i pazienti più gravi e dall’altro all’accesso in tempi più lunghi per i pazienti meno gravi, ma comunque bisognosi di cure. Nell’immediato è prevedibile che le risorse del fondo non basteranno a coprire completamente le spese previste, col rischio di determinare uno sfondamento del tetto della spesa territoriale nella maggior parte delle regioni. Il concorso dell’industria al ripiano della spesa territoriale rischia quindi di essere molto consistente. Rispetto a tale problema, l’emendamento del governo introduce la partecipazione al ripiano anche per l’industria produttrice di farmaci innovativi se il fatturato per questi supera i 300 milioni di euro. Rispetto all’accesso a questi farmaci nel medio/lungo termine, sarebbe necessario pianificare una progressiva disponibilità di risorse in modo da non depauperare il FSN, per consentire il trattamento degli altri pazienti e limitare così l’evoluzione della malattia. Va infine sottolineato che la contestuale introduzione in Italia degli altri DAA assieme a quella del sofosbuvir avrebbe potuto facilitare la competizione sui prezzi e quindi la loro riduzione, limitando questi problemi di accesso. Ci sono ragioni specifiche perché ciò non è avvenuto?
• dal punto di vista gestionale, allo stato attuale non è possibile per le strutture di governo regionale pianificare i trattamenti e la spesa e gestire al meglio i problemi legati alla mobilità attiva e passiva. Voci incontrollate si riferiscono ad una negoziazione attuata per la prima volta secondo il principio del prezzo/volume, con scaglioni di prezzo di entità via via minore. La mancanza assoluta di trasparenza nella negoziazione e l’assenza di un tavolo di confronto con le Regioni più volte richiesto nell’ambito della Conferenza Stato-Regioni alimentano tale clima di incertezza e di confusione. Se da un lato si deve riconoscere alla Gilead lo sforzo di mettere a disposizione il farmaco a prezzi differenti a seconda delle potenzialità economiche del paese acquirente, dall’altro la ribadita necessità di mantenere segreta la trattativa sul prezzo reale del farmaco cozza contro la necessità di trasparenza che è alla base di un corretto e paritario rapporto di collaborazione fra istituzioni.
Le opportunità legate ai nuovi DAA e la necessità di trasparenza
Le opportunità e le criticità che l’arrivo dei nuovi antivirali diretti stanno determinando costituiscono un buon esempio delle tensioni che si possono generare tra accesso a farmaci efficaci e incentivi all’innovazione, tra diritto alla salute e mercato. Il rischio, paradossale, che si può palesare è quello di avere a disposizione farmaci sempre più efficaci, ma sempre meno accessibili per i sistemi sanitari di tutto il mondo. Nel nostro paese queste tensioni sono state gestite con una totale assenza di trasparenza, che non ha consentito da un lato un reale e meditato confronto fra i clinici prescrittori e dall’altro una discussione pubblica (conoscendo tutti gli elementi rilevanti) su come garantire l’accesso a questi farmaci (non solo il sofosbuvir , ma anche gli altri DAA). Questo nonostante l’ordinamento italiano definisca in modo chiaro gli obblighi di trasparenza nell’ambito dell’attività amministrativa: secondo l’art. 1 della legge 241/90 (modificata e integrata dalla legge 15/2005), “L’attività amministrativa persegue i fini determinati dalla legge ed è retta da criteri di economicità, di efficacia, di pubblicità e di trasparenza, secondo le modalità previste dalla legge, nonché dai principi dell’ordinamento comunitario”. La disponibilità di informazioni, che i teorici della “concorrenza perfetta” considerano un elemento fondamentale per una (ideale) efficienza dei mercati (che producano il massimo beneficio per la società), ha una valenza non solo etica, ma anche pratica perché in questo caso faciliterebbe l’accesso ai farmaci e la gestione clinica della malattia. Ci si può solo rammaricare del fatto che la trasparenza sia un requisito di fondamentale importanza, spesso invocato, ma raramente soddisfatto.
Tabella. Valutazioni fatte in 3 Paesi europei del valore aggiunto del sofosbuvir.
| Paese | Valutazione |
| Germania (maggio 2014)10 | L’IQWiG (Institute for Quality and Efficiency in Health Care) sottolinea che il valore aggiunto del farmaco rispetto agli standard terapeutici è provato – anche se non adeguatamente quantificabile - solo per i pazienti naive con genotipo 2. Non è chiaro quanti tumori epatici possano essere prevenuti con l’uso del farmaco. |
| Francia (giugno 2014)11 | L’Haute Autorité de Santé (HAS) sostiene che può essere clinicamente appropriato l’utilizzo del farmaco in particolare in caso di epatite con cirrosi o fibrosi epatica (stadio F2-F4); nei pazienti in lista per trapianto o dopo trapianto epatico, compresi i coinfetti all’HIV, o in caso di manifestazioni exraepatiche quali il linfoma di tipo B e la crioglobulinemia mista sistemica sintomatica. L’HAS evidenzia inoltre la necessità di definire dei criteri di priorità considerando l’impatto economico del trattamento |
| Inghilterra (agosto 2014)12 | Il National Institute for Health and Clinical Excellence (NICE) pur non essendosi ancora espresso in modo definitivo sottolinea in particolare la mancanza di dati per alcuni sottogruppi di pazienti, e la mancanza di studi testa-testa che permettano di valutare direttamente il grado di innovatività rispetto agli standard terapeutici |
Si ringraziano per i suggerimenti ricevuti Giuseppe Traversa - Istituto Superiore di Sanità Francesco Nonino - Agenzia Sanitaria e Sociale Regione Emilia Romagna
Bibliografia
Data di Redazione 10/2014