L'insufficienza surrenalica primaria (morbo di Addison) è un disordine non comune (in Italia è clinicamente evidente in una persona su 8.000)1, ma importante perché cronicamente debilitante2 e suscettibile di complicanze acute, talora fatali3,4. Una volta riconosciuta, il trattamento è relativamente semplice e consente al paziente di condurre una vita normale5. La parte più difficile rimane sospettare la presenza della malattia5,6.
Stanchezza e disturbi digestivi: reperti aspecifici ma costanti
L'esordio clinico è subdolo perché i sintomi non compaiono fino a che oltre il 90% del tessuto adrenocorticale non è distrutto7. Le due ragioni che più spesso portano il paziente iposurrenalico a consultare il medico sono la stanchezza ingravescente (che può giungere ad interferire con le attività quotidiane), ed i disturbi digestivi7. Non è raro che la stanchezza venga interpretata come depressione8, o che la perdita di appetito, la nausea, i disturbi epigastrici ed il dimagrimento associato conducano alla esecuzione di una o più esofago-gastro-duodenoscopie (EGDS) non diagnostiche o solo apparentemente diagnostiche ("gastrite") e a trattamenti sintomatici non efficaci8,9. Il vomito, la diarrea ed il dolore addominale ("gastroenterite") sono meno comuni, ma possono preannunciare una crisi surrenalica5. In alcuni pazienti possono coesistere anemia o ipertransaminasemia10, 11che, se non correttamente inquadrate nell'ambito del disordine di base, rendono la diagnosi ancora più sfuggente.
Tre reperti per un primo orientamento diagnostico
Pure a fronte di sintomi così aspecifici, tre reperti, variamente associati nel singolo paziente, ma individuabili già a livello ambulatoriale, possono consentire un primo orientamento diagnostico12.
Pigmentazione. L'aspetto abbronzato della cute, espressione della attivazione dei melanociti secondaria all'eccesso di ACTH circolante è rinvenibile in oltre il 90% dei pazienti con iposurrenalismo primario (mentre è tipicamente assente nella forma secondaria da deficit ipofisario) e può, occasionalmente, precedere tutte le altre manifestazioni di malattia7. A differenza di una abbronzatura normale, la pigmentazione interessa anche aree non fotoesposte (pliche palmari, capezzoli, genitali) e talora il cavo buccale (orletto gengivale, lingua, mucosa geniena). La pigmentazione può essere del tutto assente3,8,13 ma questi casi sono rari7. La possibile coesistenza di aree di vitiligo deve rafforzare il sospetto di malattia2,14.
Ipotensione. La pressione sistolica è inferiore a 110 mmHg nel 90% dei pazienti e tende a ridursi ulteriormente spesso al di sotto di 100 e talora con manifestazioni presincopali dopo che il paziente è rimasto in posizione eretta per alcuni minuti7,12. La presenza di ipertensione rende la diagnosi di insufficienza surrenalica improbabile7. L'ipotensione esprime la ridotta responsività del cuore e dei vasi alle catecolamine per deficit di cortisolo cui si associa, nella sola forma primaria, una riduzione della volemia per deficit di aldosterone14. Il deficit di aldosterone è assente nell'iposurrenalismo secondario perché la funzione della zona glomerulare è solo parzialmente ACTH dipendente14 ed è mantenuta dalla integrità del sistema renina-angiotensina12,14. L'ipotensione può giungere ad interferire con la perfusione renale e determinare un aumento della azotemia e della creatinina14.Iposodiemia. Il sodio plasmatico è inferiore a 134 mEq/l in qualche fase della malattia nel 90% dei casi e rappresenta una delle manifestazioni biochimiche fondamentali dell'iposurrenalismo7 sia primario9 che secondario15. L'attribuzione acritica di una iposodiemia a sindrome da inappropriata secrezione di ADH (SIADH) è un evento non raro16 che può ritardare la diagnosi9. L'iposodiemia esprime l'eccesso di ormone antidiuretico (ADH) circolante per perdita del feedback negativo del cortisolo aggravato, nella sola forma primaria, dalla perdita urinaria di sodio con ritenzione di potassio per deficit di aldosterone12. L'iperkaliemia, rinvenibile nel 65% dei pazienti7, concorre con la pigmentazione ad identificare l'insufficienza surrenalica come primaria14.
Diagnosi di laboratorio: cortisolo ed ACTH
Benché i vari ormoni del corticosurrene possano essere coinvolti, la diagnosi di iposurrenalismo si basa sul dosaggio di cortisolo ed ACTH in un prelievo del mattino (quando le concentrazioni di questi ormoni sono normalmente più elevate in relazione al loro ritmo circadiano)2,12. L'assunzione preliminare di cortisonici può inibire la secrezione di ACTH ed interferire (con l'eccezione del desametasone) con il dosaggio del cortisolo endogeno7,12. Se il paziente è già in trattamento, il prelievo deve essere effettuato almeno 24 ore dopo l'ultima dose di uno steroide a breve durata di azione (idrocortisone, cortone acetato) o più a lungo in caso di steroidi a lunga durata di azione (prednisone, desametasone)7.
Cortisolo basale. Un cortisolo basale < o = 3 mcg/dl (80 nmol/L) conferma il sospetto di iposurrenalismo, mentre valori > o = 18 mcg/dl (500 nmol/L) tendono ad escluderlo2,12. Tutti i pazienti con valori intermedi necessitano di test di stimolo con ACTH esogeno12. Il trattamento con estrogeni può determinare livelli di cortisolo normale anche in presenza di iposurrenalismo per aumento della proteina legante il cortisolo (CBG)2.
Cortisolo dopo stimolo. In un paziente con sintomi, un livello di cortisolo < o = 18-20 mcg/dl (500-550 nmol/L) 30 o, preferibilmente12, 60 minuti dopo l'inoculo i.m. o e.v. di una fiala da 250 mcg di ACTH (Synacthen®) conferma la diagnosi di iposurrenalismo2,12,17. Nella maggior parte dei malati il cortisolo dopo stimolo è < o = 15 mcg/dl (415 nmol/L)17. Una risposta normale al test esclude l'insufficienza surrenalica primaria e la maggioranza dei casi di insufficienza secondaria2,12,17. Una risposta normale può non escludere un'insufficienza surrenale secondaria parziale o recente (es. entro 4 settimane da chirurgia dell'ipofisi), quando ancora non è subentrata atrofia del tessuto surrenalico secondaria al deficit di ACTH18. E' stato suggerito che un test di stimolo con 1 mcg di Synacthen® (una dose di ACTH molto più simile a quella necessaria per una stimolazione massimale del corticosurrene)12,17 possa avere maggiore sensibilità, ma i dati attualmente disponibili non sono conclusivi17 e non ovviano alla necessità di valutazioni ulteriori (test di tolleranza all'insulina, test al metirapone) di stretta competenza specialistica2.
ACTH. In presenza di ipocortisolemia basale o dopo stimolo, valori di ACTH < o = 50 pg/ml (11 pmol/L) depongono per un iposurrenalismo secondario ed indicano la necessità di concentrare l'attenzione sull'ipofisi con RM (massa? sella vuota?), studio ormonale (panipopituitarismo?) e dosaggio della ferritina (emocromatosi?)19. Nel paziente con iposurrenalismo primario (morbo di Addison) il livello di ACTH è sempre > o = 100 pg/ml (22 pmol/L)12 e spesso molto maggiore2,12,17. Il riscontro di ACTH elevato può consentire la diagnosi di iposurrenalismo primario anche in fase preclinica, quando la pigmentazione non è ancora comparsa ed il cortisolo basale e dopo stimolo possono essere normali14. L'approccio ulteriore al morbo di Addison varia tra il paziente oncologico ed il paziente non-oncologico. I pazienti con metastasi surrenali bilaterali hanno in genere neoplasie disseminate e non pongono problemi diagnostici20. Nel paziente non oncologico occorre distinguere tra adrenalite autoimmune e forme non autoimmuni, principalmente adrenalite TBC ed adrenoleucodistrofia12.
Adrenalite Autoimmune
L'adrenalite autoimmune (distruzione progressiva del corticosurrene per infiltrazione centripeta da parte di linfociti citotossici) rappresenta oltre l'80% dei casi di iposurrenalismo primario nel nostro paese14. La malattia può essere sospettata clinicamente per la frequente coesistenza di vitiligo e/o altri disordini autoimmuni nel paziente o nei familiari21, ma la conferma richiede il riscontro nel siero di anticorpi diretti contro la corticale del surrene (ACA) e/o contro enzimi coinvolti nella steroidogenesi, in particolare la 21-idrossilasi (21-OH)1,22. La presenza simultanea di ACA ed anti 21-OH consente una diagnosi definitiva di adrenalite autoimmune, ma la diagnosi può basarsi sul riscontro di un solo anticorpo se il titolo è elevato1. Questi anticorpi sono presenti nel siero in oltre il 90% dei pazienti con malattia all'esordio1,22, ma tendono a negativizzarsi e dopo 15 anni di malattia gli ACA sono positivi nel 10% dei pazienti e gli anti 21-OH nel 60% circa1,22. Nei familiari asintomatici il valore predittivo degli ACA e degli anticorpi anti-21OH per lo sviluppo di insufficienza surrenale clinica è molto maggiore nel bambino (100%) che nell'adulto (20-30%)14. I principali esami da richiedere dopo diagnosi di adrenalite autoimmune sono riportati in Tabella 1.
Adrenalite Tubercolare
La distruzione tubercolare dei surreni rappresenta il 13% circa dei casi di morbo di Addison in Italia1, ed è la causa più comune di morbo di Addison in India e nei paesi in via di sviluppo27. La malattia deve essere sospettata in presenza di febbre e/o anomalie al radiogramma del torace, ma il coinvolgimento surrenalico può essere la sola espressione di malattia27,28. La TC mostra spesso surreni ingranditi (a differenza che nell'adrenalite autoimmune), talora con predominanza di lato27, che nel corso degli anni tendono a contrarsi e calcificano nel 50% dei casi29. Quando i surreni sono ridotti di volume e le calcificazioni sono assenti, la diagnosi differenziale radiologica da surreni con adrenalite autoimmune può non essere possibile20. Il trattamento può essere seguito dal recupero della funzione surrenalica30, ma ciò è raro31,32. La terapia con rifampicina accelera il metabolismo del cortisolo e richiede terapia sostitutiva a dosaggio più alto32. Altre cause di adrenalite infettiva sono inusuali, ma occorre considerare la possibilità di infezione da HIV. Benché in uno studio condotto su 222 pazienti italiani con insufficienza surrenalica non si siano riscontrati pazienti sieropositivi1, l'infezione da HIV è un fattore di rischio ben noto per insufficienza surrenalica, perché i surreni possono essere distrutti da infezioni opportunistiche (Citomegalovirus, TBC) e/o essere inibiti funzionalmente dai numerosi farmaci (es. ketoconazolo-Nizoral®) usati in questi pazienti33.
Adrenoleucodistrofia
La adrenoleucodistrofia deve essere considerata in presenza di iposurrenalismo primario non autoimmune non tubercolare in un maschio in età pediatrica od adulto-giovanile7. Il disordine, trasmesso con meccanismo ereditario legato al cromosoma X, è probabilmente meno raro di quanto ritenuto in passato34. La malattia è caratterizzata dall'accumulo di acidi grassi a catena molto lunga (VLCFA) nel SNC e nel surrene7. L'insufficienza surrenalica può precedere la paraparesi e le altre manifestazioni neurologiche, ed in una minoranza dei malati costituire la sola manifestazione clinica7. La diagnosi richiede il dosaggio dei VLCFA (Very Long Chain Fatty Acids) nel siero, da inviarsi a laboratori specializzati (es. Istituto Neurologico Nazionale "Carlo Besta" - Milano)34. Il riconoscimento di un caso indice può condurre alla dimostrazione di casi subclinici tra i familiari34,35.
Terapia
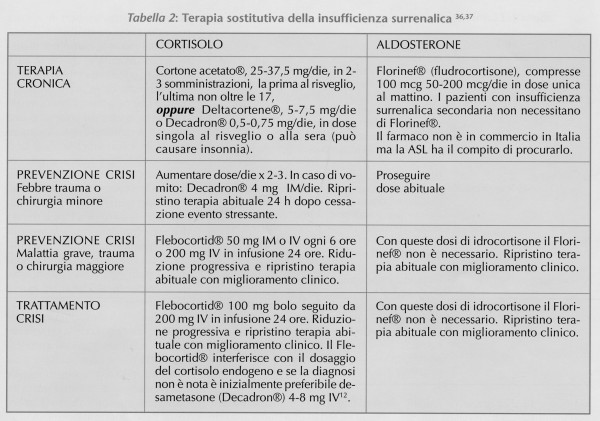
La terapia sostitutiva della insufficienza surrenalica si basa sul trattamento del deficit di cortisolo e nella grande maggioranza dei pazienti con forma primaria - di aldosterone (Tabella 2)36,37. Se la astenia fisica e sessuale persistono nonostante questo trattamento, un beneficio ulteriore può derivare dalla correzione del deficit androgenico con deidro-epiandrosterone (DEA), specie nella donna, in cui la zona reticolare del surrene è la principale fonte di androgeni38. Questo trattamento, tuttavia, non è ancora parte della routine clinica36,37. L'istruzione del malato, come nel diabete, è fondamentale, specie in caso di vomito o malattia intercorrente39 ed il paziente deve avere un documento che ne attesti l'iposurrenalismo in caso che un trauma o una malattia lo rendano improvvisamente inabile a comunicare2,12. Il Cortone acetato®, il farmaco più comunemente usato in Italia, ha una emivita limitata ad 80 minuti7 ed alcuni esperti preferiscono un trattamento con steroidi a lunga durata di azione (prednisone, desametasone), ritenendo che questi garantiscano un livello di cortisolo ematico più stabile7,40. Non esistono studi di confonto affidabili tra questi due approcci e la risposta è spesso individuale41. Il dosaggio ottimale ed il monitoraggio della terapia nel singolo paziente sono un compromesso tra teoria e pratica42,43,44 il cui obiettivo mira alla regressione delle manifestazioni di malattia senza induzione di ipercortisolismo (aumento di peso, ipertensione, osteopenia) e/o eccesso mineralcorticoide (edemi perimalleolari, ipokaliemia)7. L'uso del livello di ACTH come guida alla terapia sostitutiva è controverso2 e può condurre ad eccesso di trattamento2,12,37, mentre il dosaggio della attiità reninica plasmatica (PRA) è utile nel trattamento sostitutivo aldosteronico con Florinef®2. Altri metodi di monitoraggio (es.curve del cortisolo) sono poco utilizzati nella pratica36,37.
Insufficienza surrenalica acuta (crisi surrenalica)
Quando l'iposurrenalismo è noto, la causa più comune di insufficienza acuta è la mancata assunzione della terapia (es. vomito) o il mancato adeguamento di dosaggio in risposta ad uno stress46 (Tabella 2). Più spesso, la crisi consegue ad un iposurrenalismo primario cronico non riconosciuto2,12, e può essere precipitata da uno stress intercorrente (es. infezione, trauma, chirurgia) o dalla somministrazione di farmaci (es. tiroxina-Eutirox®) che accelerano il catabolismo del cortisolo residuo47,48. Più raramente, una emorragia surrenale bilaterale può causare iposurrenalismo acuto d'esordio49; in questi casi, in assenza di un fattore causale definito (es. setticemia o terapia anticoagulante), va sempre esclusa una sindrome da anticorpi antifosfolipidi49,50. La crisi surrenalica spesso non viene sospettata, perché i sintomi sono aspecifici2 e l'iposodiemia e l'iperkaliemia possono essere mascherate dal vomito, dalla diarrea e dai liquidi infusi7. Il dolore addominale può simulare un addome acuto chirurgico51. La presenza di ipotensione, specie se refrattaria alla espansione di volume o alle catecolamine deve sempre sollevare il sospetto di possibile crisi surrenalica52 che, se non riconosciuta, può essere fatale3,4. Il sospetto deve essere seguito da un prelievo per cortisolo ed ACTH (in qualunque ora del giorno)12 iniziando il trattamento (Tabella 2) prima dell'esito di laboratorio52,53,54. Valori di cortisolo < 15 mcg/dl (415 nmol/L) sono chiaramente inappropriati in corso di stress9 e confermano l'iposurrenalismo. Pure in assenza di un valore soglia universalmente accettato2,12, un livello di cortisolo > o = 34 mcg/dl (938 nmol/L) consente di escludere con buona probabilità l'insufficienza surrenalica e di sospendere lo steroide52. Pazienti con valori intermedi tra 15 e 34 mcg/dl (415-938 nmol/L) debbono essere sottoposti a test con Synacthen®. I pazienti in cui il cortisolo dopo stimolo aumenta meno di 9 mcg/dl (248 nmol/L) debbono essere considerati iposurrenalici e proseguire il trattamento per 5-11 giorni52,55, mentre quelli con aumento superiore è improbabile che ne derivino beneficio e possono sospenderlo52. Nel paziente critico, specie se con shock settico, il test al Synacthen® può essere gravato da falsi positivi e va ripetuto una volta che il paziente è stabile prima di confermare la necessità di terapia cronica56.
Gravidanza
Al giorno d'oggi la maggior parte delle donne adeguatamente trattate sono in grado di affrontare gravidanza e parto senza rischi57 ma prima dell'introduzione della terapia steroidea la gravidanza nelle pazienti con malattia di Addison era rara e gravata da elevata morbidità materno-fetale58. Se non già nota, la diagnosi in gravidanza può essere difficile perché stanchezza, vomito, pigmentazione ed ipotensione possono accompagnarsi ad una gravidanza per sé59, ed il livello di cortisolo può essere apparentemente normale per l'aumento dei livelli sierici di proteina legante il cortisolo (CBG) legati all'iperestrogenismo gravidico57,58. La pigmentazione buccale è tuttavia assente in una gravidanza normale57, e la risposta allo stimolo con Synacthen® ed il livello di ACTH sono analoghi a quelli della donna iposurrenalica non gravida57,58. Il trattamento è sovrapponibile a quello al di fuori dello stato gravidico (Tabella 2), ma nel terzo trimestre è spesso necessario aumentare i dosaggi di glucocorticoide eFlorinef® sulla base dei sintomi e del potassio37. L'uso della attività reninica plasmatica come guida alla terapia con Florinef®, è consigliato da alcuni esperti57, ma spesso di difficile interpretazione per l'aumento fisiologico legato alla gravidanza2,37. Il travaglio ed il parto (specie se cesareo) richiedono una profilassi della crisi come per le procedure chirurgiche (Tabella 2).
Bibliografia 1. Falorni A et al. Italian Addison Network Study: Update on diagnostic criteria for the etiological classification of primary adrenal insufficiency. J Clin Endocr Metab 2004; 89: 1598. 2. Arit W et al. Adrenal Insufficiency. Lancet 2003; 361: 1881. 3. Brosnan CM et al. Addison's disease. BMJ 1996; 312: 1085. 4. Ringstad J et al. Rapidly fatal Addison's disease : three case reports. J Intern Med 1991; 230: 465. 5. Ten S et al. Addison's disease. J Clin Endocrinol Met 2001; 86: 2909. 6. Paterson JR et al. Delayed diagnosis of Addison's disease. Ann Clin Biochem 1990; 27: 378. 7. Orth Dn et al. The Adrenal Cortex. In Williams Textbook of Endocrinology (9th ed.), Saunders 1998 : 517. 8. Keljo DJ et al. Just in time. New Engl J Med 1996; 334: 46. 9. Smith JC et al. Misinterpretation of serum cortisol in a patient with hyponatraemia. BMJ 2004; 328: 215. 10. Massari M et al. Le alterazioni delle transaminasi (1° parte). Inf Farm 2001; n. 2-3: 74. 11. Roblin X et al.Unexplained hypertransaminasemia: a clue to the diagnosis of Addison's disease. Eur J Gastroent Hepatol 2003; 15: 929. 12. Oelkers W. Adrenal Insufficency. New Engl J Med 1996; 335: 1209. 13. Kendersesky A et al. White Addison's disease : what is the possible cause ? J Endocrinol Invest 1999; 22: 395. 14. Betterle C. Malattia di Addison. In Betterle C, Le Malattie Autoimmuni, Piccin 2001: 136. 15. Chanson P. Severe hyponatremia as a frequent revealing sign of hypopituitarism after 60 years of age. Eur J Endocrinol 2003; 6: 177. 16. Saeed BO et al. Severe hyponatraemia: investigation and management in a district general hospital. J Clin Pathol 2002; 55: 893. 17. Dorin RI et al. Diagnosis of adrenal insufficiency. Ann Intern Med 2003; 139: 194. 18. Streeten DHP et al. The potential for serious consequences from misinterpreting normal responses to the rapid adrenocorticotropin test. J Clin Endocrinol Met 1996; 81: 285. 19. Wahid S et a. Clinical picture: the pituitary gland and haemochromatosis. Lancet 2001; 357: 115. 20. Vita JA et al. Clinical clues to the cause of Addison's disease. Am J Med 1985; 78: 461. 21. Eisenbarth GS et al. Autoimmune polyendocrine syndromes. New Engl J Med 2004; 350: 2068. 22. Falorni A et al. Autoantibodies in autoimmune polyendocrine syndrome type II. Endocr Met Clin North Am2002; 31: 369. 23. Armstrong L et al. Addison's disease presenting as reduced insulin requirement in insulin dependent diabetes. BMJ 1996; 312: 1601. 24. Schatz DA et al. Autoimmune polyglandular syndrome type II: clinical syndrome and treatment. Endocr Met Clin North Am 2002; 31: 320. 25. Perheentupa J. APS1-APECED: clinical disease and therapy. Endocr Met Clin North Am 2002; 31: 339. 26. O'Leary C et al. Coeliac disease and autoimmune Addison's disease: a clinical pitfall. Quarterly J Med 2002;95: 79. 27. Serter R et al. Acute adrenal crisis together with unilateral adrenal mass caused by isolated TB of adrenal gland. Endocr Pract 2003; 9: 157. 28. Llewelyn M et al. Acute adrenal insufficiency precipitated by isolated involvement of the adrenal gland by tuberculosis. J Infect 1999; 39: 244. 29. Villabona CM et al. Tuberculous Addison's disease: utility of CT in diagnosis and follow-up. Eur J Radiol 1993;17: 210. 30. Penrice J et al. Recovery of adrenocortical function following treatment of tuberculous Addison's disease.Postgrad Med J 1992; 68: 204. 31. Bathia E et al. Tuberculous Addison's dsease: lack of normalization of adrenocortical function after antituberculous chemotherapy. Clin Endocrinol 1998; 48: 35. 32. Kaven K et al. Adrenal function during tuberculous infection and effects of antituberculous treatment on endogenous and exogenous steroids. Int J Tuberc Lung Dis 1998; 35 : 419. 33. Sellmeyer De et al. Endocrine and metabolic disturbances in human immunodeficiency virus infection and the acquired immunodeficiency syndrome. Endocr Rev 1996; 17: 518. 34. Laureti S et al. X-linked adrenoleukodystrophy is a frequent cause of idiopathic Addison's disease in young adult male patients. J Clin Endocr Met 1996; 81: 470. 35. Ronghe MD et al. The importance of testing for adrenoleukodystrophy in males with idiopathic Addison's disease. Arch Dis Child 2002; 86: 185. 36. Jeffcoate W.Assessment of corticosteroid replacement therapy in adults with adrenal insufficiency. Ann Clin Biochem 1999; 36: 11. 37. Oelkers W. Therapeutic strategies in adrenal insufficiency. Ann Endocrin (Paris) 2001;62: 212. 38. PJ Hunt et al. Improvement in mood and fatigue after DHEA replacement in Addison' disease. JCEM 2000;85: 4650. 39. Peacey SR et al. Corticosteroid therapy and intercurrent illness: the need for continued patient education.Postgrad Med J 1993; 69: 282. 40. Coursin DB et al. Corticosteroid supplementation for adrenal insufficiency. JAMA 2002; 287: 236. 41. Schimmer BP et al. Adrenocortical steroids: therapeutic uses. In: Goodman & Gilman's The Pharmacologic Basis of Therapeutics (McGraw-Hill) 2001: 1668. 42. Peacey Sr et al. Glucocorticoid replacement therapy: are patients overtreated and does it matter ? Clin Endocrinol 1997; 46: 255. 43. Howlett TA et al. An assessment of optimal hydrocortisone replacement therapy. Clin Endocrinol 1997; 46: 263. 44. Romijn JA et al.Intrinsic imperfections of endocrine replacement therapy. Eur J Endocrinol 2003; 149: 91. 45. Anonimo.Trattamento sostitutivo della insufficienza surrenalica. Boll Inf Farm 1999 (1): 35. 46. Krasner AS. Glucocorticoid-induced adrenal insufficiency. JAMA 1999; 282: 671. 47. Murray JS et al. Deterioration of symptoms after starting thyroxine. BMJ 2001; 323: 332. 48. Graves L et al. Addisonian crisis precipitated by thyroxine therapy: a complication of type 2 autoimmune polyglandular syndrome. South Med J 2003; 8: 824-827. 49. Vella A et al. Adrenal hemorrhage: a 25-year experience at the Mayo Clinic. Mayo Clin Proc 2001; 76: 161. 50. Espinosa G et al. Adrenal involvement in the antiphospholipid syndrome: clinical and immunologic characteristics of 86 patients. Medicine 2003; 82: 106. 51. Laws SAM et al. Adrenal insufficiency masquerading as acute abdomen. Hosp Med 2001; 62: 118. 52. Cooper M et al. Corticosteroid insufficiency in acutely ill patients. New Engl J Med 2003; 348: 727. 53. Minneci PC et al. Meta-analysis : the effect of steroids on survival and shock during sepsis depends on dose.Ann Intern Med 2004; 141: 47. 54. Luce JM. Physicians should administer low-dose corticosteroids selectively to septic patients until an ongoing trial is completed. Ann Intern Med 2004; 141: 70. 55. Annane D et al. Corticosteroids for severe sepsis and septic shock : a systematic review and meta-analysis.BMJ 2004; 329: 480. 56. Briegel J et al.A comparison of the adrenocortical response during septic shock and after complete recovery.Intens Care Med 1996; 22: 894. 57. Ambrosi B et al.Diagnosis and management of Addison's disease during pregnancy. J Endocrinol Invest2003; 7: 698. 58. Garner PR.Disorders of the adrenal cortex in pregnancy. In Burrow and Duffy, Medical Complications during Pregnancy (Saunders), 1999: 171. 59. Gradden C et al. Clinical picture: Addison's disease in pregnancy. Lancet 2001; 357: 1197.


{kind=link}
{kind=link}