

L'ipotesi che ha guidato le scelte di una rilettura complessiva di un problema che è senza dubbio di competenza strettamente specialistica, sia a livello di diagnosi, che di scelte terapeutiche, che di gestione assistenziale può essere così formulata:
1. Si tratta di una patologia che da ormai vent'anni rappresenta, allo stesso tempo, un ambito di rilevante importanza dal punto di vista del carico assistenziale diagnostico–terapeutico e dei relativi costi, ed una domanda aperta in termini del grado di evidenze clinico–epidemiologiche sulla efficacia–effettività dei trattamenti a disposizione. Questi sono sostanzialmente centrati su interferoni, ma con una importante frazione di pazienti esposte/i ad altri trattamenti, senza che ci siano criteri certi per la scelta tra le diverse strategie, né dal punto di vista diagnostico né dell'efficacia–effettività. Da notare inoltre – ed il problema non è banale – che ad essere trattata è soprattutto una delle espressioni della malattia, quella relapsing–remitting, che riguarda all'esordio l'80% della popolazione, ma che progressivamente si riduce man mano che la malattia–disabilità progredisce.
2. Prima ancora che il paradigma della Health Technology Assessment assumesse il ruolo attuale di metodologia ufficiale nella valutazione del ruolo complessivo e specifico degli interventi ad alto costo e di profilo incerto di beneficio/costo, la Sclerosi Multipla (SM) è stata uno dei capitoli più controversi, specificamente in Gran Bretagna, nel rapporto tra la logica valutativa del NICE e le scelte concrete di rimborsabilità da parte dei sistemi pubblici e/o delle coperture assicurative.
3. All'"innovazione" introdotta (dopo anni di monopolio da parte degli interferoni) con il natalizumab (registrato e commercializzato con una domanda esplicitamente aperta sulla sua sicurezza) lo scenario è cambiato in modo apparentemente sostanziale con l'arrivo quasi concomitante di due farmaci che spostano ulteriormente il meccanismo ipotizzato di target terapeutico, e permettono di affrontare una terapia di così lungo periodo con un trattamento orale.
4. I risultati sui quali si basano i nuovi farmaci sono stati prodotti con trial che ripropongono in modo più esplicito la domanda sulla reale efficacia del trattamento "storico" con interferone.
5. Mentre la SM passa da patologia con un profilo di semi–orfanità terapeutica a patologia con più opzioni, ci si accorge che lungo tutti questi anni non si sono resi disponibili (con pochissime eccezioni) studi "robusti" in termini di "outcome effectiveness" sufficienti a documentare se e quanto l'intensità diagnostico–terapeutica produca di fatto un miglioramento sostanziale sulla vita, e non solo sulle scale che ne quantificano le espressioni sintomatico– funzionali, con strumenti riconosciuti come insoddisfacenti.
6. Pur essendo competenza (proprietà?) degli specialisti, le/i pazienti con SM sono una realtà ben presente e visibile, sia nella società che nella pratica di medicina generale. Rappresentano di fatto in un certo senso il paradigma più chiaro di problemi con i quali "si convive" in attesa che la medicina produca qualcosa di certo–definitivo, ma per i quali non sono disponibili risorse paragonabili a quelle delle tecnologie diagnostico–terapeutiche, per assicurare una presa in carico assistenziale rivolta alla vita di queste/i pazienti, che più diventano "gravi", più si fanno invisibili all'attenzione medica: le loro storie sono note solo ai soggetti che ne sono i protagonisti, alle associazioni che li seguono, a caregiver e badanti, senza che possano assumere una visibilità epidemiologica ed assistenziale nella "grande letteratura".
7. Lo scenario così delineato (e per il quale la letteratura, al di là di quella utilizzata in questa revisione, è disponibile su richiesta) non è molto diverso da quello che caratterizza altre aree neurologico–comportamentali come la demenza ed i suoi trattamenti "specifici", o i problemi comportamentali dell'anziano che sono oggetto (altrettanto controverso in termini di efficienza–effettività–sicurezza) di trattamenti studiati e registrati per patologie psichiatriche.
8. La revisione che si propone mira dunque a fornire più che una informazione dettagliata su tutto quanto dice la letteratura (che si assume sarà disponibile a livello specialistico con tutte le sue [non]–[controverse] "evidenze"), un percorso di metodologia e di riflessione che può essere di interesse più generale, in quanto tocca il problema:
- del "che fare" quando la EBM ufficiale include strutturalmente l'incertezza; - del "che dire" alle/ai pazienti, e all'opinione pubblica, quando l'assistenza deve essere qualificata e vissuta– praticata, per essere responsabile–etica, come una ricerca nel pieno senso della parola.
Logica e struttura di questa revisione–riflessione Si è scelto di lasciare quanto più possibile lo spazio al "vedere", riducendo al massimo i commenti e le parole. La letteratura più recente è riletta a partire dalle pubblicazioni (prevalentemente in riviste non specialistiche ad altissimo Impact Factor) che hanno fatto il punto conoscitivo e/o hanno documentato le innovazioni terapeutiche, e/o hanno fatto il punto applicativo dei risultati innovativi. Si sono scelte le immagini che si ritengono più sintetiche e "rappresentative" integrate con testi che mirano a favorirne la lettura, ma anche a guidare al riconoscimento della fondatezza dei punti formulati nella ipotesi. Si spera che chi è interessato possa così meglio formulare un giudizio proprio, così da arrivare alle conclusioni con la possibilità e l'impegno di confrontare un punto di vista da "lettura di un aggiornamento", e quello di chi è direttamente coinvolto nella produzione–ricerca di risposte clinicamente ed epidemiologicamente [più] soddisfacenti.
Per un inquadramento generale
1. La/e causa/e della SM sono tuttora sconosciute nonostante l'abbondanza di studi epidemiologici, genetici, di biologia cellulare e molecolare che si sono succeduti ed articolati via via che si rendevano disponibili le diverse tecnologie. Il consenso sul fatto che ci sia una interazione tra fattori ambientali e profili genetici complessi coincide di fatto con una affermazione assolutamente generale che può/deve applicarsi sostanzialmente a tutte le patologie per le quali non sia nota una causa monogenica.
2. C'è anche un accordo di massima sui meccanismi di base coinvolti nello sviluppo della patologia che viene considerata una malattia infiammatoria del nevrasse, cronica ed invalidante, che si sviluppa a partire da infiltrazioni linfocitarie che danneggiano la mielina e gli assoni del cervello e midollo spinale. Non è difficile peraltro riconoscere che un quadro così definito è compatibile con uno spettro molto ampio di fattori più o meno specifici, ciò che si traduce nella diversità dei target terapeutici dei vecchi e nuovi farmaci che sono stati proposti 1.
3. Il processo diagnostico via via precisato negli ultimi anni, fino alla sua ultima formulazione (Figura 1) 2 sintetizza bene, nella sua complessa articolazione, la problematicità di un inquadramento in grado di essere non solo descrittivo, ma predittivo-prognostico.
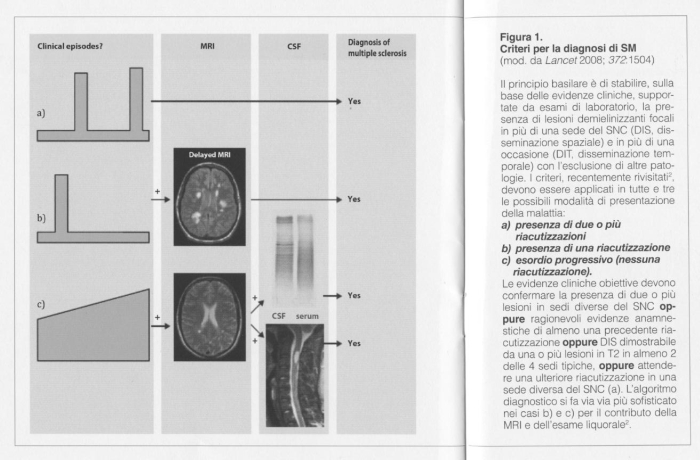
4. La epidemiologia della "storia naturale" della SM non può che essere il risultato dei tanti livelli di approssimazione che si sono definiti sopra. La fase precoce della malattia è caratterizzata da ricorrenti episodi di disfunzione neurologica abitualmente reversibili (SM-RR, con riacutizzazione e remissione). Con l'inevitabile progredire della malattia il quadro patologico dominante è caratterizzato da diffusi processi di attivazione della microglia associati a estesi fenomeni di neurodegenerazione il cui correlato clinico è costituito dalla crescente disabilità 1. Nella maggior parte delle/dei pazienti la malattia si presenta come episodio acuto di deficit neurologico mono-focale denominata Sindrome Clinicamente Isolata (SCI). La successiva occorrenza di un secondo episodio in sede diversa dal precedente consente di formulare una diagnosi clinicamente definita di SM-RR. La frequenza di riacutizzazioni della malattia è molto variabile (ma raramente eccede 1-2 episodi per anno) e condiziona fortemente il tempo necessario alla formulazione della diagnosi. Il supporto di esami strumentali e di laboratorio in grado di dimostrare:
a) presenza e distribuzione di lesioni infiammatorie e/o degenerazione assonale (Risonanza Magnetica Nucleare),
b) interferenza con la velocità di conduzione di stimoli lungo specifiche vie sensitive o sensoriali (potenziali evocati),
c) sintesi intratecale di anticorpi oligoclonali (esame del liquor cerebrospinale) contribuiscono al percorso diagnostico secondo criteri standardizzati. L'incompleto recupero dopo ripetute riacutizzazioni giustifica la persistenza di quadri neurologici invalidanti a cui si aggiunge, in circa il 65% dei pazienti, l'ingresso in una fase cosiddetta di progressione secondaria (SM-PS, progressiva secondaria).
Nel 20% circa dei pazienti la malattia si presenta in forma progressiva sin dall'esordio (SM-PP, progressiva primaria) 1. Con una età media di esordio di circa 30 anni, una aspettativa di vita intorno ai 65 anni, ed una mediana di sopravvivenza di circa 30 anni dall'esordio, la SM si caratterizza come malattia cronica di lunga durata in grado di interferire pesantemente e per diverse decadi nella vita delle persone affette 1,3.
5. Ancor più difficile, evidentemente, una valutazione della epidemiologia assistenziale della SM, che permetta di incorporare e tener conto criticamente della variabilità dei percorsi diagnostici e terapeutici e dei criteri di utilizzazione degli strumenti che misurano la progressione delle diverse espressioni, oggettive e funzionali, della patologia.
6. I farmaci, tra loro molto diversi, che sono stati registrati come capaci di modificare il decorso di malattia (DMD, disease modifying drugs), fanno riferimento (rimandando a meccanismi diversi) ad attività "immunomodulatoria" (una volta ancora la genericità del termine dice bene la approssimazione del loro profilo farmacologico). I DMD sono attualmente utilizzati con
a) buona efficacia nella riduzione di frequenza di nuovi episodi nelle forme SM-RR,
b) nessuna efficacia su deficit neurologici stabilizzati,
c) con ancora discutibile evidenza di efficacia nel prevenire la progressione di malattia e la crescente invalidità a lungo termine.
Nonostante forti raccomandazioni all'uso sempre più precoce di terapie immunomodulanti permangono ancora incertezze sulla durata di tali terapie per ottenere un ragionevole rapportorischi/benefici. Gli studi clinici controllati su tutti gli attuali e futuri DMD nella SM-RR hanno durate di 2-3 anni ovvero considerevolmente più brevi di ciò che accade nella pratica clinica.Tutto ciò è oggi particolarmente evidente se si considerano che alcuni pazienti con SMRR risultano trattati in modo continuativo sin dal 1993, anno di approvazione dell'interferone ß-1b 3.Ancora poco è conosciuto sugli effetti di interruzione, dopo anni, del trattamento con immunomodulanti e sul possibile rischio di sviluppare maggiore disabilità 4. La recente pubblicazione dei risultati di 4 importanti RCT con l'attesissimo avvento delle prime formulazioni orali di Disease-modifying drugs non costituiscono solo una nuova opzioneterapeutica, ma aprono una nuova fase nel trattamento della SM 5 riproponendo formalmente i temi sempre aperti di:
Il "punto di vista" dei nuovi trial Come anticipato nella introduzione, la scelta di "far vedere" i risultati (studi FREEDOMS 6, TRANSFORMS 7, CLARITY 8, TRANSFORMS estensione 9), limitandosi a mettere in evidenza i dati principali, vuole stimolare un'attenzione che si ponga esplicitamente il problema della [più o meno grande] comparabilità dei profili di efficacia e dei rapporti tra differenze clinicamente e statisticamente significative. Un commento sugli aspetti di safety (che avrebbero bisogno di un approfondimento a parte) è proposto in modo sintetico in un paragrafo "complessivo" a parte.
Studio FREEDOMS 6
A Placebo-Controlled Trial of Oral Fingolimod in Relapsing Multiple Sclerosis
E' importante soltanto sottolineare la specificità del contenuto informativo dei singoli grafici in termini di 'misure di esito'. Il tempo intercorrente tra inizio del trattamento e prima ricaduta è evidentemente molto significativo, ma dice poco sulla evoluzione di lungo periodo (A).Anche la riduzione della progressione di malattia è "misurata" su periodi brevi e per variazioni di Expanded Disability Status Scale (EDSS) piccole (0,5-1 punto) (B).Il miglior controllo sulla attività di malattia nei pazienti trattati è ben documentato con una misura di esito "surrogata" quale è l'assenza di lesioni gadolinio (+) alla MRI (C).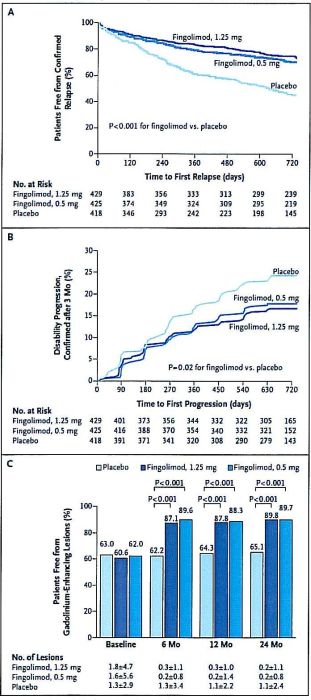
Studio TRANSFORMS 7
Oral Fingolimod or Intramuscular Interferon for Relapsing Multiple Sclerosis
La riduzione della frequenza di riacutizzazioni e l'incremento dell'intervallo di tempo libero da riacutizzazioni misurano ovviamente lo stesso fenomeno e si confermano la misura di esito più sensibile per valutare differenze di efficacia tra trattamenti ma rimangono poco informativi sulla evoluzione o progressione della malattia.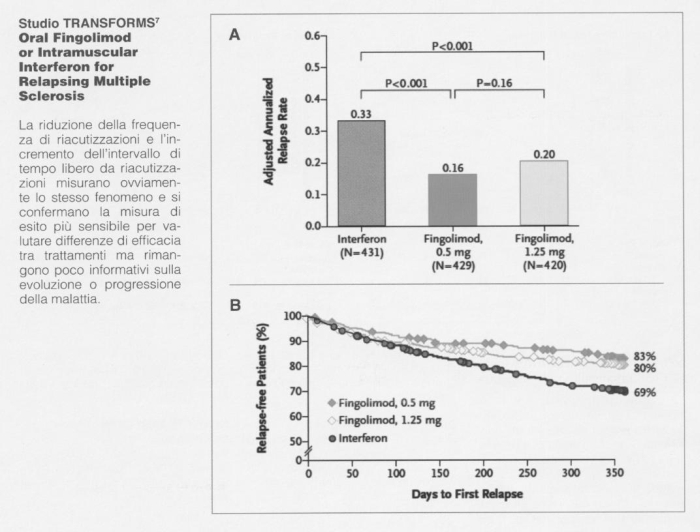
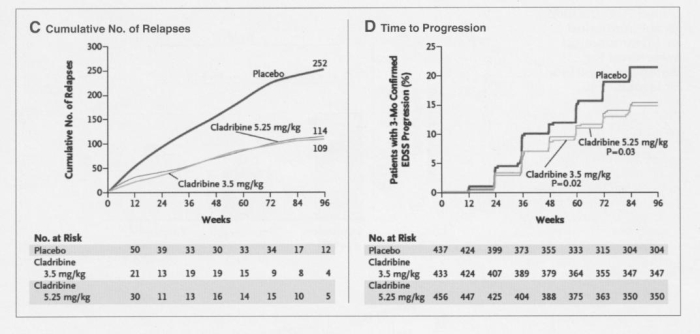
Studio CLARITY 8
A Placebo-Controlled Trial of Oral Cladribine for Relapsing Multiple Sclerosis
I riquadri A – B – C illustrano in modo diverso la stessa misura di esito precedentemente descritta, mentre il quadro D confrontabile con quello mostrato per lo studio FREEDOMS mostra una analoga riduzione nella progressione di malattia per tutti i pazienti trattati vs placebo.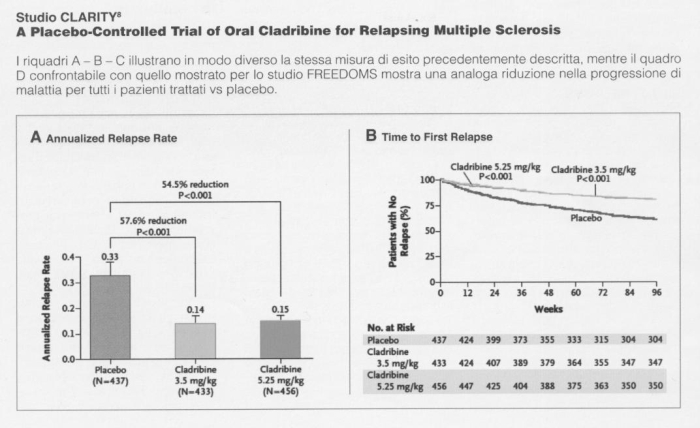
Alcuni aspetti di safety
La ricorrenza di "eventi avversi" è riportata con frequenza molto alta in tutti i gruppi di tutti i trials considerati. Per un eventuale approfondimento e per visionare gli elenchi particolareggiati e completi si rimanda ai lavori in originale. Qui si è scelto di non trascurare gli eventi ritenuti più seri per i quali è già necessaria una farmacosorveglianza 'attiva' e che ci aiuti, come metodo e come 'sguardo', a riportare l'attenzione sul paziente trattato piuttosto che sul farmaco.
Studio FREEDOMS
Effetti indesiderati gravi sono stati riportati nel 10,1% dei pazienti trattati con fingolimod 0,5 mg, nell'11,9% di quelli trattati con fingolimod 1,25 mg e nel 13,4% di quelli trattati con placebo. I più frequenti, ciascuno osservato in otto pazienti, sono consistiti in bradicardia, recidiva della sclerosi multipla e carcinoma a cellule basali. Tutti gli altri eventi avversi gravi si sono manifestati al massimo in quattro pazienti (<1%) per gruppo. I sette episodi di bradicardia (di cui sei asintomatici) nei pazienti trattati con fingolimod (quattro nel gruppo 0,5 mg e tre nel gruppo 1,25 mg) sono emersi durante il periodo di monitoraggio successivo alla somministrazione della prima dose. Nel corso dello studio si sono verificati tre decessi, due con placebo, uno con fingolimod 1,25 mg. Nel gruppo placebo, le cause dei decessi sono state embolia polmonare e incidente stradale, nel gruppo fingolimod suicidio.
Studio TRANSFORMS
Due casi di infezione mortale si sono verificati nel gruppo trattato con la dose di 1,25 mg di fingolimod: uno di varicella zoster primariamente disseminata, uno di encefalite da herpes simplex. Altri effetti indesiderati occorsi nei pazienti trattati con fingolimod sono stati infezioni non fatali da herpes virus, bradicardia e blocco atrioventricolare, ipertensione arteriosa, edema maculare, carcinoma cutaneo e aumento degli enzimi epatici.
Studio CLARITY
Nei pazienti trattati con cladribina, gli effetti indesiderati più frequenti sono stati linfocitopenia (21,6% nel gruppo 3,5 mg and 31,5% nel gruppo 5,25 mg vs 1,8% nel gruppo placebo) ed herpes zoster (rispettivamente in 8 e 12 pazienti vs nessun paziente). L'incidenza di eventi avversi gravi è stata dell'8,4% nel gruppo cladribina 3,5 mg, del 9,0% nel gruppo cladribina 5,25 mg e del 6,4% nel gruppo placebo. Le infezioni o le infestazioni sono state riportate come gravi rispettivamente nel 2,3%, 2,9% e 1,6% dei pazienti. L'herpes zoster è risultato grave in tre pazienti trattati con cladribina (due nel gruppo 5,25 mg). Le neoplasie (benigne, maligne o di natura non specificata) sono state riportate tra gli eventi avversi gravi nell'1,4% dei pazienti nel gruppo cladribina 3,5 mg e nello 0,9% del gruppo 5,25 mg, rispetto a nessun caso osservato nei pazienti trattati con placebo.
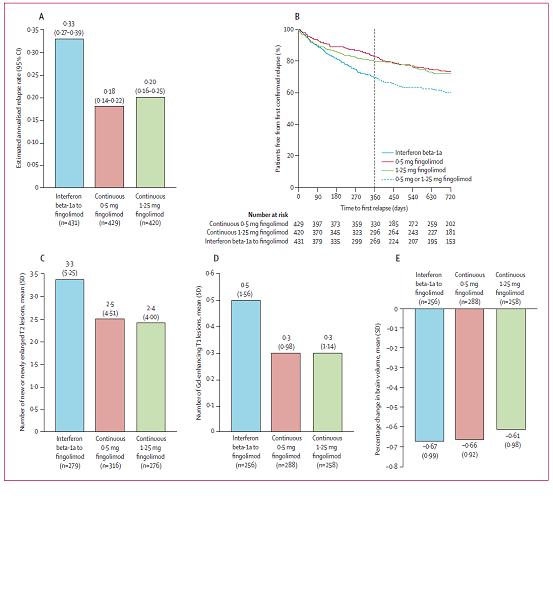
Studio TRANSFORMS estensione
Il passaggio da interferon beta-1a a fingolimod ha comportato un aumento dell'efficacia senza problemi inattesi di sicurezza.
Studio TRANSFORMS estensione 9
Comparison of fingolimod with interferon beta-1a in relapsing-remitting multiple sclerosis: a randomised extension of the TRANSFORMS study.
Il nuovo trattamento (Fingolimod) vs trattamento standard (Interferon beta-1a) conferma la propria efficacia sulle stesse misure di esito (numero di riacutizzazioni e lesioni alla MRI) anche nella fase di estensione dello TRANSFORMS proponendosi perciò come farmaco di prima linea.
Un commento complessivo
-Tutti gli studi esaminati hanno utilizzato come end point primario la frequenza annualizzata di riacutizzazione della malattia (SM-RR). Basse o alte dosi di fingolimod e cladribina si sono dimostrate più efficaci sia del placebo che dell'interferone beta-1a. Tale superiorità rispetto ai comparator è emersa anche su end point secondari quali lesione alla RMI e tempo di progressione della disabilità valutata come incremento di 0,5-1 punto (confermato dopo 3 mesi) alla Expanded Disability Status Scale (EDSS).
-Eventi "avversi" sono stati riportati dettagliatamente e con grande frequenza sia per il gruppo trattato a basse o alte dosi di farmaco che, inaspettatamente per il lettore, anche per il gruppo placebo. Tali eventi sono stati classificati in maggioranza come lievi e moderati e spesso indistinguibili da eventi clinici propri della malattia. Permangono significativi effetti indesiderati gravi la cui rilevanza ha verosimilmente condizionato l'approvazione delle diverse agenzie regolatorie.
-Fingolimod è stato approvato come terapia di prima linea in USA, Svizzera ed Australia, mentre Canada e European Medicines Agency (EMA) hanno limitato l'indicazione a pazienti non responder a farmaci di prima linea (interferone e/o glatiramer acetato) o a casi di malattia grave. Cladribina è stata approvata come terapia di prima linea in Russia ed Australia, ma respinta dalla Food and Drug Administration (FDA) statunitense e dall'EMA principalmente per problemi di sicurezza a lungo termine, alla luce dei dati di incidenza di neoplasie dello studio di fase 3 CLARITY10.
-Programmi di sorveglianza a lungo termine per nuove terapie "disease-modifying" nella SM sono oggi ritenuti di particolare importanza anche e soprattutto in considerazione della breve durata (2 anni) degli studi clinici controllati di fase 3. La Leucoencefalopatia Progressiva Multifocale (PML), evento avverso "raro" e "grave", verificato in pazienti con SM in trattamento con natalizumab, è emerso solo dopo l'approvazione del farmaco e la sua reale incidenza appare oggi incrementare verosimilmente in correlazione con trattamenti di più lunga durata.
-In particolare i risultati della fase di estensione dello studio TRANSFORMS hanno diretta rilevanza per la pratica clinica proprio in considerazione della indicazione all'uso di fingolimod come trattamento di seconda linea. La fase di estensione dello studio appare molto prossima alla concreta possibilità per molti pazienti in trattamento con interferone beta 1a o glatiramer acetato (somministrabili solo per via i.m o s.c.) di transitare alla nuova terapia (per via orale) con un apparente ed accettabile profilo di rischio/beneficio 11. Lo studio di estensione, come modello e metodologia, è una stretta approssimazione di pratica clinica e fornisce utili informazioni o anticipazioni su tollerabilità, sicurezza ed efficacia. Ma, come nota cautelativa, il rischio globale o complessivo sulle nuove immunoterapie nella SM è ancora sconosciuto. Dati di farmacovigilanza, programmi di risk managment, promozione di registri di patologia capaci di raccogliere dati su base storica e di popolazione in contesti reali di assistenza sono, oggi, più che mai necessari per più approfondite e complete analisi di rischio/beneficio (a breve ma, soprattutto, a lungo termine) che non potrebbero mai essere condotte sulla base dei soli controlled clinical trials 11.
- Il contributo informativo dei Registri di patologia, sul tema della efficacia e sicurezza a lungo termine, è ampiamente esplorato nel supplemento del gennaio 2011 di Neurology 3,12-17 per comprendere quali benefici o limitazioni possano derivare da studi a lungo termine sulle diverse opzioni terapeutiche nella SM e soprattutto quali misure di outcomes si dimostrano veramente rilevanti e consigliabili per clinical trials e futuri registri 3.
-Nonostante le rilevanti differenze nei Registri di SM (presenti in qualche caso da diverse decadi in Europa, Canada e Stati Uniti) alcune evidenze su mortalità e su pattern e fattori predittivi di progressione della disabilità appaiono sostanzialmente confermate 13.
-Obiettivi principali nel trattamento della SM sono la riduzione dei sintomi e della progressione di malattia. Dati osservazionali dei Registri di malattia indicano come la progressione della disabilità avvenga lentamente nel corso di decadi. Perciò, studi di follow up a lungo o lunghissimo termine sono necessari per comprendere anche l'impatto di terapie disease modifying nella SM. Sulla base dei dati attualmente disponibili da studi di Long-Term Follow Up (LTFU) alcune raccomandazioni possono essere formulate proprio per orientare e migliorare studi futuri. Il raggiungimento di livelli stabili di EDSS di 4 o di 6 possono essere considerati "milestones" sufficientemente affidabili perché, una volta raggiunti, la malattia progredisce inesorabilmente con modalità prevedibili. Il raggiungimento di livelli stabili di EDSS 6, può essere raccomandato come endpoint centrale in registri di patologia per la possibilità di essere rilevato più precocemente rispetto al dato di mortalità che, come abbiamo già evidenziato, è molto più tardivo nella lunga storia di una malattia come la SM 12,13,15.
-Aperto rimane il problema dell'assenza di un gruppo controllo o di un comparator in studi a lungo termine ed appare del tutto evidente che solo combinando differenti tipi di studi è possibile raggiungere evidenze capaci di fornire risposte su
-In ultimo appare opportuno rilevare che sebbene fingolimod (e altri farmaci già approvati dalla agenzie regolatorie) sia efficace nel trattamento della SM con riacutizzazioni e remissioni,ancora nulla appare per le forme più gravi di malattia SM con progressione sia secondaria che primaria.
1. Compston A, Coles A. Seminar: Multiple sclerosis. Lancet 2008; 372:1502-17.
2. Polman CH et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol 2011; 69:292-302.
3. Bates D. Introduction: Long-term outcomes in patients with multiple sclerosis. Neurology 2011; 76 (Suppl 1): S1-S2.
4. Ford CC et al. A prospective open-label study of glatiramer acetate: over a decade of continuous use in multiple sclerosis patients. Mult Scler 2006; 12:309-20.
5. Carroll WM. Editorial: Oral Therapy for Multiple Sclerosis — Sea Change or Incremental Step? N Engl J Med 2010; 362:456-8.
6. Kappos L et al. for the FREEDOMS Study Group. A placebo-controlled trial of oral fingolimod in Relapsing Multiple Sclerosis. N Engl J Med 2010; 362:387-401.
7. Cohen JA et al. for the TRANSFORMS Study Group. Oral Fingolimod or Intramuscular Interferon for Relapsing Multiple Sclerosis. N Engl J Med 2010; 362:402-15.
8. Giovannoni G et al. for the CLARITY Study Group, A Placebo-Controlled Trial of Oral Cladribine for Relapsing Multiple Sclerosis. N Engl J Med 2010; 362:416-26.
9. Khatri B et al. on behalf of the TRANSFORMS Study Group. Comparison of fingolimod with interferon beta-1a in relapsing-remitting multiple sclerosis: a randomised extension of the TRANSFORMS study. Lancet Neurol 2011; 10:520-9.
10. Editorial: Balancing the benefits and risks of new drugs for MS. Lancet Neurol 2011; 10:491.
11. Kieseier BC, Wiendl H. Comment: Transforming multiple sclerosis trials into practical reality. Lancet Neurol 2011; 10:493-4.
12. Hurwitz BJ. Registry studies of long-term multiple sclerosis outcomes: description of key registries. Neurology 2011; 76 (Suppl 1): S3-S6.
13. Hurwitz BJ. Analysis of current multiple sclerosis registries. Neurology 2011; 76 (Suppl 1): S7-S13.
14. Bates D. Treatment effects of immunomodulatory therapies at different stages of multiple sclerosis in short-term trials. Neurology 2011; 76 (Suppl 1): S14-S25.
15. Freedman M. Long-term follow-up of clinical trials of multiple sclerosis therapies. Neurology 2011; 76 (Suppl 1): S26-S34.
16. Freedman M. Improving long-term follow-up studies of immunomodulatory therapies. Neurology 2011; 76 (Suppl 1): S35-S38.
17. Bates D. Summary: registry data. Neurology 2011; 76 (Suppl 1): S39-S41.
Data di Redazione 04/2011